Cobimetinib
Chemical formula: C₂₁H₂₁F₃IN₃O₂ Molecular mass: 531.318 g/mol PubChem compound: 16222096
Interactions
Cobimetinib interacts in the following cases:
OATP1B1 substrates, OATP1B3 substrates, OCT1 substrates
In vitro studies show that cobimetinib is not a substrate of the liver uptake transporters OATP1B1, OATP1B3 and OCT1, however, it weakly inhibits these transporters. The clinical relevance of these findings has not been investigated.
BCRP substrates
In vitro, cobimetinib is a moderate inhibitor of BCRP (Breast Cancer Resistance Protein). No clinical DDI studies have been conducted to assess this finding, and clinically relevant inhibition of intestinal BCRP cannot be ruled out.
CYP1A2 substrates
In vitro, cobimetinib is a potential inducer of CYP1A2 and may therefore reduce the exposure of substrates of this enzyme e.g., theophylline. No clinical DDI studies have been conducted to assess the clinical relevance of this finding.
CYP3A inducers
Co-administration of cobimetinib with a strong CYP3A inducer was not assessed in a clinical study, however, a reduction in cobimetinib exposure is likely. Therefore, concomitant use of moderate and strong CYP3A inducers (e.g. carbamazepine, rifampicin, phenytoin, and St. John's Wort) should be avoided. Alternative agents with no or minimal CYP3A induction should be considered. Given that cobimetinib concentrations are likely to be significantly reduced when co-administered with moderate to strong CYP3A inducers, patient's efficacy may be compromised.
P-glycoprotein inhibitors
Cobimetinib is a substrate of P-glycoprotein (P-gp). Concomitant administration of P-gp inhibitors such as ciclosporin and verapamil may have the potential to increase plasma concentrations of cobimetinib.
Strong CYP3A inhibitors
Cobimetinib is metabolized by CYP3A and cobimetinib AUC increased approximately 7 fold in the presence of a strong CYP3A inhibitor (itraconazole) in healthy subjects. The magnitude of interaction could potentially be lower in patients.
Strong CYP3A inhibitors: Avoid concurrent use of strong CYP3A inhibitors during treatment with cobimetinib. Strong CYP3A inhibitors include, but are not limited to ritonavir, cobicistat, telaprevir, lopinavir, itraconazole, voriconazole, clarithromycin, telithromycin, posaconazole, nefazodone and grapefruit juice. If concomitant use of a strong CYP3A inhibitor is unavoidable, patients should be carefully monitored for safety. For strong CYP3A inhibitors used short-term (7 days or less), consider interrupting cobimetinib therapy during the duration of inhibitor use.
Moderate CYP3A inhibitors
Caution should be exercised if cobimetinib is coadministered with moderate CYP3A inhibitors. Moderate CYP3A inhibitors include, but are not limited to, amiodarone, erythromycin, fluconazole, miconazole, diltiazem, verapamil, delavirdine, amprenavir, fosamprenavir, imatinib. When cobimetinib is co-administered with a moderate CYP3A inhibitor, patients should be carefully monitored for safety.
Severe renal impairment
There are minimal data for cobimetinib in patients with severe renal impairment, therefore an effect cannot be excluded. Cobimetinib should be used with caution in patients with severe renal impairment.
Severe hepatic impairment
Patients with severe hepatic impairment may have increased plasma concentrations of unbound cobimetinib compared to patients with normal hepatic function. Liver laboratory abnormalities can occur with cobimetinib and caution should be used in patients with any degree of hepatic impairment.
Haemorrhage
Haemorrhagic events, including major haemorrhagic events can occur. Caution should be used in patients with additional risk factors for bleeding, such as brain metastases, and/or in patients that use concomitant medications that increase the risk of bleeding (including antiplatelet or anticoagulant therapy).
Left ventricular dysfunction
Decrease in LVEF from baseline has been reported in patients receiving cobimetinib. Median time to initial onset of events was 4 months (1-13 months).
LVEF should be evaluated before initiation of treatment to establish baseline values, then after the first month of treatment and at least every 3 months or as clinically indicated until treatment discontinuation. Decrease in LVEF from baseline can be managed using treatment interruption, dose reduction or with treatment discontinuation.
All patients restarting treatment with a dose reduction of cobimetinib should have LVEF measurements taken after approximately 2 weeks, 4 weeks, 10 weeks and 16 weeks, and then as clinically indicated.
Patients with a baseline LVEF either below institutional lower limit of normal (LLN) or below 50% have not been studied.
Dose modification advice for left ventricular dysfunction
Permanent discontinuation of cobimetinib treatment should be considered if cardiac symptoms are attributed to cobimetinib and do not improve after temporary interruption.
Recommended dose modifications for cobimetinib in patients with left ventricular ejection fraction (LVEF) decrease from baseline:
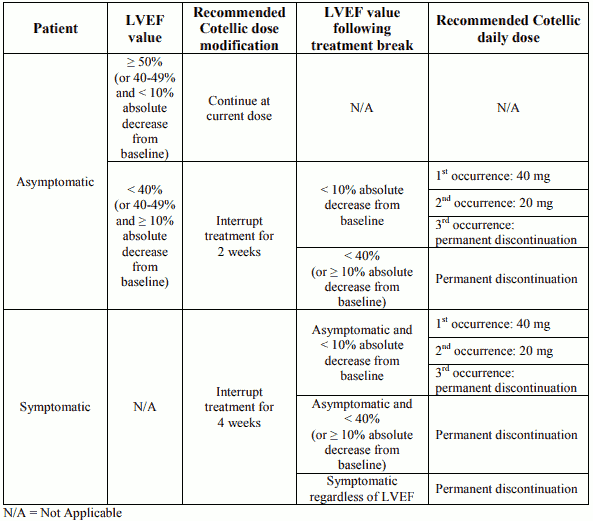
Vemurafenib treatment can be continued when cobimetinib treatment is modified, if clinically indicated.
Liver laboratory abnormalities
Liver laboratory abnormalities can occur when cobimetinib is used in combination with vemurafenib and with vemurafenib as a single agent.
Liver laboratory abnormalities, specifically increases in Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST), and Alkaline Phosphatase (ALP), have been observed in patients treated with cobimetinib plus vemurafenib.
Liver value abnormalities should be monitored by liver laboratory tests before initiation of combination treatment and monthly during treatment, or more frequently as clinically indicated.
Grade 3 liver laboratory abnormalities should be managed with vemurafenib treatment interruption or dose reduction. Manage Grade 4 liver laboratory abnormalities with treatment interruption, dose reduction or with treatment discontinuation of both cobimetinib and vemurafenib.
For Grade 1 and 2 liver laboratory abnormalities, cobimetinib and vemurafenib should be continued at the prescribed dose.
Grade 3: Cobimetinib should be continued at the prescribed dose. The dose of vemurafenib may be reduced as clinically appropriate. Please refer to the vemurafenib SmPC.
Grade 4: Cobimetinib treatment and vemurafenib treatment should be interrupted. If liver laboratory abnormalities improve to Grade ≤1 within 4 weeks, cobimetinib should be restarted at a dose reduced by 20 mg and vemurafenib at a clinically appropriate dose, per its SmPC.
Cobimetinib treatment and vemurafenib treatment should be discontinued if liver laboratory abnormalities do not resolve to Grade ≤1 within 4 weeks or if Grade 4 liver laboratory abnormalities recur after initial improvement.
Rash
Rash events may occur with either cobimetinib or vemurafenib treatment. The dose of cobimetinib and/or vemurafenib may be either temporarily interrupted and/or reduced as clinically indicated.
Additionally, for:
Grade ≤2 (tolerable) rash should be managed with supportive care. Cobimetinib dosing can be continued without modification.
Grade 2 (intolerable) or Grade ≥3 acneiform rash: General dose modification recommendations in Table 1 for cobimetinib should be followed. Vemurafenib dosing can be continued when cobimetinib treatment is modified (if clinically indicated).
Grade 2 (intolerable) or Grade ≥3 non-acneiform or maculopapular rash: Cobimetinib dosing can be continued without modification if clinically indicated. Vemurafenib dosing may be either temporarily interrupted and/or reduced, please refer to its SmPC for further information.
Serous retinopathy
Serous retinopathy (fluid accumulation within the layers of the retina) has been observed in patients treated with MEK-inhibitors, including cobimetinib. The majority of events were reported as chorioretinopathy or retinal detachment.
Median time to initial onset of serous retinopathy events was 1 month (range 0-9 months). Most events observed in clinical trials were resolved, or improved to asymptomatic Grade 1, following dose interruption or reduction.
Patients should be assessed at each visit for symptoms of new or worsening visual disturbances. If symptoms of new or worsening visual disturbances are identified, an ophthalmologic examination is recommended. If serous retinopathy is diagnosed, cobimetinib treatment should be withheld until visual symptoms improve to Grade ≤1. Serous retinopathy can be managed with treatment interruption, dose reduction or with treatment discontinuation.
Diarrhoea
Cases of Grade ≥3 and serious diarrhoea have been reported in patients treated with cobimetinib. Diarrhoea should be managed with anti-diarrhoeal agents and supportive care. For Grade ≥3 diarrhoea that occurs despite supportive care, cobimetinib and vemurafenib should be withheld until diarrhoea has improved to Grade ≤1. If Grade ≥3 diarrhoea recurs, the dose of cobimetinib and vemurafenib should be reduced.
Photosensitivity
Grade ≤2 (tolerable) photosensitivity should be managed with supportive care.
Grade 2 (intolerable) or Grade ≥3 photosensitivity: Cobimetinib and vemurafenib should be interrupted until resolution to Grade ≤1. Treatment can be restarted with no change in cobimetinib dose. Vemurafenib dosing should be reduced as clinically appropriate.
Rhabdomyolysis, CPK elevations
Rhabdomyolysis has been reported in patients receiving cobimetinib.
If rhabdomyolysis is diagnosed, cobimetinib treatment should be interrupted and CPK levels and other symptoms monitored until resolution. Depending on the severity of rhabdomyolysis, dose reduction or treatment discontinuation may be required.
Grade 3 and 4 CPK elevations, including asymptomatic elevations over baseline, also occurred in patients receiving cobimetinib with vemurafenib in clinical trials. The median time to first occurrence of Grade 3 or 4 CPK elevations was 16 days (range: 11 days to 10 months); the median time to complete resolution was 16 days (range: 2 days to 15 months).
Serum CPK and creatinine levels should be measured before initiation of treatment, to establish baseline values, and then monitored monthly during treatment, or as clinically indicated. If serum CPK is elevated, check for signs and symptoms of rhabdomyolysis or other causes. Depending on the severity of symptoms or CPK elevation; treatment interruption, dose reduction or treatment discontinuation may be required.
Dose modification advice for rhabdomyolysis and Creatine phosphokinase (CPK) elevations
Rhabdomyolysis or symptomatic CPK elevations
Cobimetinib treatment should be interrupted. If rhabdomyolysis or symptomatic CPK elevations do not improve within 4 weeks, cobimetinib treatment should be permanently discontinued. If severity is improved by at least one grade within 4 weeks, cobimetinib could be restarted at a dose reduced by 20 mg, if clinically indicated. Patients should be closely monitored. Vemurafenib dosing can be continued when cobimetinib treatment is modified.
Asymptomatic CPK elevations
Grade 4: Cobimetinib treatment should be interrupted. If CPK elevations do not improve to Grade ≤3 within 4 weeks following dose interruption, cobimetinib treatment should be permanently discontinued.
If CPK improves to Grade ≤3 within 4 weeks, cobimetinib could be restarted, if clinically indicated, at a dose reduced by 20 mg and the patient should be closely monitored. Vemurafenib dosing can be continued when cobimetinib treatment is modified.
Grade ≤3: After rhabdomyolysis has been ruled out, cobimetinib dosing does not need to be modified.
Pregnancy
There are no data from the use of cobimetinib in pregnant women. Studies in animals have shown embryolethality and foetal malformations of the great vessels and skull. Cobimetinib should not be used during pregnancy unless clearly necessary and after a careful consideration of the needs of the mother and the risk to the foetus.
Nursing mothers
It is not known whether cobimetinib is excreted in human breast milk. A risk to the newborns/infants cannot be excluded. A decision should be made whether to discontinue breast-feeding or discontinue cobimetinib therapy, taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
Carcinogenesis, mutagenesis and fertility
Women of childbearing potential
Women of childbearing potential should be advised to use two effective contraceptive methods, such as a condom or other barrier method (with spermicide, if available) during treatment with cobimetinib and for at least three months following treatment discontinuation.
Fertility
There are no data in humans for cobimetinib. In animals, no fertility studies have been performed, but adverse effects were seen on reproductive organs. The clinical relevance of this is unknown.
Effects on ability to drive and use machines
Cobimetinib has minor influence on the ability to drive or use machines. Visual disturbances have been reported in some patients treated with cobimetinib during clinical trials. Patients should be advised not to drive or use machines if they experience visual disturbances or any other adverse effects that may affect their ability.
Adverse reactions
Summary of the safety profile
The safety of cobimetinib in combination with vemurafenib has been evaluated in 247 patients with advanced BRAF V600 mutated melanoma in Study GO28141.The median time to onset for the first Grade ≥3 adverse events was 0.6 months in the cobimetinib plus vemurafenib arm vs 0.8 months in the placebo plus vemurafenib arm.
The safety of cobimetinib in combination with vemurafenib has also been evaluated in 129 patients with advanced BRAF V600 mutated melanoma in Study NO25395. The safety profile of Study NO25395 was consistent with that observed in Study GO28141.
In Study GO28141, the most common adverse reactions (>20%) observed with a higher frequency in the cobimetinib plus vemurafenib arm were diarrhoea, rash, nausea, pyrexia, photosensitivity reaction, increased alanine aminotransferase, increased aspartate aminotransferase, increased blood creatine phosphokinase, and vomiting. The most common adverse reactions (>20%) observed with a higher frequency in the placebo plus vemurafenib arm were arthralgia, alopecia, and hyperkeratosis. Fatigue was observed at similar frequencies in both arms.
Tabulated list of adverse reactions
ADRs are based on results from a multi-centre, randomised, double-blind, placebo-controlled, Phase III Study (GO28141) that evaluated the safety and efficacy of cobimetinib in combination with vemurafenib as compared to vemurafenib alone in previously untreated BRAF V600 mutation-positive patients with unresectable locally advanced (Stage IIIc) or metastatic melanoma (Stage IV).
ADR frequencies are based upon the safety analysis of patients treated with cobimetinib plus vemurafenib with a median follow up of 11.2 months (data cut-off date of 19 September 2014).
ADRs which were reported in melanoma patients are listed below by MedDRA body system organ class, frequency and grade of severity. The following convention has been used for the classification of frequency: Very common ≥1/10, Common ≥1/100 to <1/10, Uncommon ≥1/1,000 to <1/100, Rare ≥1/10,000 to <1/1,000, Very rare <1/10,000
The following list provides adverse reactions considered associated with the use of cobimetinib. Within each frequency grouping, ADRs are presented in order of decreasing severity and were reported according to NCICTCAE v 4.0 (common toxicity criteria) for assessment of toxicity in Study GO28141.
Adverse drug reactions in patients treated with cobimetinib in combination with vemurafenib in Study GO28141^:
Neoplasms benign, malignant and unspecified (incl. cysts and polyps)
Common: Basal cell carcinoma, Cutaneous squamous cell carcinoma**, Keratoacanthoma**
Blood and lymphatic system disorders
Very Common: Anaemia
Metabolism and nutrition disorders
Common: Dehydration, Hypophosphataemia, Hyponatremia, Hyperglycaemia
Eye disorders
Very Common: Serous retinopathya, Blurred vision
Common: Visual impairment
Vascular disorders
Very Common: Hypertension, Haemorrhage*
Respiratory, thoracic and mediastinal disorders
Common: Pneumonitis
Gastrointestinal disorders
Very Common: Diarrhoea, Nausea, Vomiting
Skin and subcutaneous tissue disorders
Very Common: Photosensitivityb, Rash, Rash maculo-papular, Dermatitis acneiform, Hyperkeratosis**
Musculoskeletal and connective tissue disorders
Uncommon: Rhabdomyolysis***
General disorders and administration site conditions
Very Common: Pyrexia, Chills
Investigations
Very Common: Blood CPK increased, ALT increased, AST increased, Gamma-Glutamyltransferase (GGT) increased, Blood ALP increased
Common: Ejection fraction decreased, Blood bilirubin increased
^ Data cut-off date of 19 September 2014
* Please refer to the paragraph Haemorrhage in the "Description of selected adverse reactions" section
** Please refer to the paragraph Cutaneous squamous cell carcinoma, keratoacanthoma and hyperkeratosis in the "Description of selected adverse reactions" section.
*** Please refer to the paragraph Rhabdomyolysis in the "Description of selected adverse reactions" section.
a Includes both chorioretinopathy and retinal detachment events indicative of serous retinopathy
b Combined figure includes reports of photosensitivity reaction, sunburn, solar dermatitis, actinic elastosis
Description of selected adverse reactions
Haemorrhage
Bleeding events have been reported more frequently in the cobimetinib plus vemurafenib arm than in the placebo plus vemurafenib arm (all types and Grades: 13% vs 7%). The median time to first onset was 6.1 months in the cobimetinib plus vemurafenib arm.
The majority of events were Grade 1 or 2 and non-serious. Most events resolved with no change in cobimetinib dose. Major haemorrhagic events (including intracranial and gastrointestinal tract haemorrhage) were reported in the post-marketing setting. The risk of haemorrhage may be increased with concomitant use of antiplatelet or anticoagulant therapy. If haemorrhage occurs, treat as clinically indicated.
Rhabdomyolysis
Rhabdomyolysis has been reported in the post-marketing setting. Signs or symptoms of rhabdomyolysis warrant an appropriate clinical evaluation and treatment as indicated, along with cobimetinib dose modification or discontinuation according to the severity of the adverse reaction.
Photosensitivity
Photosensitivity has been observed with a higher frequency in the cobimetinib plus vemurafenib arm vs placebo plus vemurafenib arm (47% vs 35%). The majority of events were Grades 1 or 2, with Grade ≥3 events occurring in 4% of patients in the cobimetinib plus vemurafenib arm vs 0% in the placebo plus vemurafenib arm.
There were no apparent trends in the time of onset of Grade ≥3 events. Grade ≥3 photosensitivity events in the cobimetinib plus vemurafenib arm were treated with primary topical medicinal products in conjunction with dose interruptions of both cobimetinib and vemurafenib.
No evidence of phototoxicity was observed with cobimetinib as a single agent.
Cutaneous squamous cell carcinoma, keratoacanthoma and hyperkeratosis
Cutaneous squamous cell carcinoma has been reported with a lower frequency in the cobimetinib plus vemurafenib arm vs placebo plus vemurafenib arm (all Grade: 3% vs 13%). Keratoacanthoma has been reported with a lower frequency in the cobimetinib plus vemurafenib arm vs placebo plus vemurafenib arm (all Grade: 2% vs 9%). Hyperkeratosis has been reported with a lower frequency in the cobimetinib plus vemurafenib vs placebo plus vemurafenib arm (all Grade: 11% vs 30%).
Serous retinopathy
Cases of serous retinopathy have been reported in patients treated with cobimetinib. For patients reporting new or worsening visual disturbances, an ophthalmologic examination is recommended. Serous retinopathy can be managed with treatment interruption, dose reduction or with treatment discontinuation.
Left ventricular dysfunction
Decrease in LVEF from baseline has been reported in patients receiving cobimetinib. LVEF should be evaluated before initiation of treatment to establish baseline values, then after the first month of treatment and at least every 3 months or as clinically indicated until treatment discontinuation. Decrease in LVEF from baseline can be managed using treatment interruption, dose reduction or with treatment discontinuation.
Laboratory abnormalities
Liver laboratory abnormalities
Liver laboratory abnormalities, specifically ALT, AST, and ALP have been observed in patients treated with cobimetinib in combination with vemurafenib. Liver laboratory tests should be monitored before initiation of combination treatment and monthly during treatment, or more frequently if clinically indicated.
Blood creatine phosphokinase increase
Asymptomatic increases in blood CPK levels were observed with a higher frequency in the cobimetinib plus vemurafenib arm vs placebo plus vemurafenib arm in Study GO28141. One event of rhabdomyolysis was observed in each treatment arm of the study with concurrent increases in blood CPK.
Table 4 provides the frequency of measured liver laboratory abnormalities and elevated creatine phosphokinase for all Grades and Grades 3-4.
Table 4. Liver function and other laboratory tests observed in the Phase III Study GO28141:
| Changes in reported laboratory data | Cobimetinib plus Vemurafenib (n=247) (%) | Placebo plus Vemurafenib (n=246) (%) | ||
|---|---|---|---|---|
| All Grades | Grades 3-4 | All Grades | Grades 3-4 | |
| Liver function test | ||||
| Increased ALP | 69 | 7 | 55 | 3 |
| Increased ALT | 67 | 11 | 54 | 5 |
| Increased AST | 71 | 7 | 43 | 2 |
| Increased GGT | 62 | 20 | 59 | 17 |
| Increased blood bilirubin | 33 | 2 | 43 | 1 |
| Other laboratory abnormalities | ||||
| Increased blood CPK | 70 | 12 | 14 | <1 |
Special populations
Elderly patients
In the Phase III study with cobimetinib in combination with vemurafenib in patients with unresectable or metastatic melanoma (n=247), 183 patients (74%) were <65 years of age, and 44 patients (18%) were 65-74 years of age, 16 (6%) were 75-84 years of age, and 4 patients (2%) were aged >85 years. The proportion of patients experiencing adverse events (AE) was similar in the patients aged <65 years and those aged >65 years. Patients ≥65 years were more likely to experience serious adverse events (SAEs) and experience AEs leading to discontinuation of cobimetinib than those <65 years.
Renal impairment
No pharmacokinetic trial in subjects with renal impairment has been conducted. Dose adjustment is not recommended for mild to moderate renal impairment based on the results of the population pharmacokinetic analysis. There are minimal data for cobimetinib in patients with severe renal impairment. Cobimetinib should be used with caution in patients with severe renal impairment.
Hepatic impairment
No dose adjustment is recommended in patients with hepatic impairment.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.