Flibanserin
Chemical formula: C₂₀H₂₁F₃N₄O Molecular mass: 390.402 g/mol PubChem compound: 6918248
Mechanism of action
The mechanism of action of flibanserin in the treatment of premenopausal women with hypoactive sexual desire disorder is not known.
Pharmacodynamic properties
Receptor Binding
In vitro, flibanserin demonstrated high affinity for the following serotonin (5-hydroxytryptamine or 5-HT) receptors: agonist activity at 5-HT1A and antagonist activity at 5-HT2A. Flibanserin also has moderate antagonist activities at the 5-HT2B, 5-HT2C, and dopamine D4 receptors.
Cardiac Electrophysiology
The effect of flibanserin on the QT interval was evaluated in a randomized, double-blind, placebo- and active- (single dose moxifloxacin) controlled crossover study in 56 healthy men and women. Subjects in the flibanserin groups received either 50 mg twice a day (equivalent to the daily recommended dosage) or 100 mg three times a day (3 times the daily recommended dosage) administered for 5 days. The time frame for electrocardiogram (ECG) measurements covered maximum plasma concentrations of flibanserin and relevant metabolites. In this study, flibanserin did not prolong the QT interval to any clinically relevant extent. The mean increase in heart rate associated with the 100 mg three times a day dose of flibanserin compared to placebo ranged from 1.7 to 3.2 beats per minute.
Pharmacokinetic properties
Flibanserin showed dose-proportional pharmacokinetics for Cmax after single oral doses of 100 mg to 250 mg (the recommended and 2.5 times the recommended dosage, respectively) in healthy female subjects. Steady state was achieved after 3 days of dosing. The extent of exposure (AUC0-∞) with once-daily dosing of 100 mg of flibanserin was increased 1.4-fold as compared to a single dose.
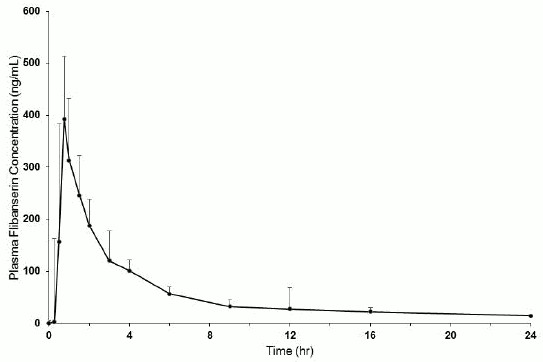
Figure 1. Mean + SD Plasma Flibanserin Concentration-Time Profiles in Healthy Female Subjects Following a Single Oral Dose of 100 mg of Flibanserin (Linear Scale):
Absorption
Following oral administration of a single 100 mg dose of flibanserin in healthy premenopausal women (N=8), mean (SD) Cmax was 419 (206) ng/mL and mean (SD) AUC0-inf was 1543 (511) ng*hr/mL. Median (range) time to reach Cmax was 0.75 (0.75 to 4.0) hours. Absolute bioavailability of flibanserin following oral dosing is 33%.
Effect of Food
Food increased the extent of absorption and slowed the rate of absorption of a 50 mg dose of flibanserin (one half the recommended dosage). Low-, moderate-, and high-fat meals increased flibanserin AUC0-inf by 1.18-, 1.43-, and 1.56-fold; increased Cmax by 1.02-, 1.13-, and 1.15-fold; and prolonged median Tmax to 1.5, 0.9, 1.8 hours from 0.8 hours under fasted conditions, respectively.
Distribution
Approximately 98% of flibanserin is bound to human serum proteins, mainly to albumin.
Elimination
Metabolism
Flibanserin is primarily metabolized by CYP3A4 and, to a lesser extent, by CYP2C19. Based on in vitro and/or in vivo data, CYP1A2, CYP2B6, CYP2C8, CYP2C9, and CYP2D6 contribute minimally to the metabolism of flibanserin. After a single oral solution dose of 50 mg 14C-radiolabeled flibanserin, 44% of the total 14C-flibanserin related radioactivity was recovered in urine, and 51% was recovered in feces. Flibanserin is extensively metabolized to at least 35 metabolites, most of them occurring in low concentrations in plasma. Two metabolites could be characterized that showed plasma concentrations similar to that achieved with flibanserin: 6,21-dihydroxy-flibanserin-6,21-disulfate and 6-hydroxy-flibanserin-6-sulfate. These two metabolites are inactive.
Excretion
Flibanserin has a mean terminal half-life of approximately 11 hours.
Specific Populations
Hepatic Impairment
Single 50 mg oral doses of flibanserin were administered to 10 patients with mild hepatic impairment (Child-Pugh score of 6 points), 4 patients with moderate hepatic impairment (Child-Pugh score of 8-9 points), and 14 healthy subjects matched by age, weight, and gender. Systemic flibanserin exposure (AUC0-inf) increased 4.5-fold in patients with mild hepatic impairment, compared to subjects with normal hepatic function, and t1/2 was longer (26 hours compared to 10 hours in matching healthy controls). Due to the small number of patients (n=4) with moderate hepatic impairment enrolled in the study, it is not possible to make conclusions about the quantitative effect of moderate hepatic impairment on flibanserin exposure. Flibanserin is contraindicated in patients with hepatic impairment.
Renal Impairment
Single 50 mg oral doses of flibanserin were administered to 7 patients with mild to moderate renal impairment (GFR 30 to 80 mL/min), 9 patients with severe renal impairment (GFR <30 mL/min, not on dialysis), and 16 healthy subjects matched by age, weight, and gender. Flibanserin exposure (AUC0-inf) increased 1.1-fold in patients with mild to moderate renal impairment and 1.2-fold in patients with severe renal impairment, compared to the healthy control subjects.
Race/Ethnicity
A cross-study comparison between healthy Japanese women and Caucasian women with HSDD showed that flibanserin exposure was approximately 1.4-fold higher in Japanese women. When the mean flibanserin exposure in Japanese women was adjusted for weight, the AUCtau,ss in Japanese women was 2246 ng*hr/mL, which is comparable to 2080 ng*hr/mL in Caucasian women. The similarity in weight-adjusted AUCtau,ss suggests that weight, not race, is the factor contributing to the observed difference in flibanserin exposure between Japanese and Caucasian women.
Age
No formal study has been conducted to study the effect of age on flibanserin exposures.
Drug Interaction Studies
Drugs that Increase Flibanserin Exposure
The effects of other drugs on the pharmacokinetics of flibanserin are presented in Table 1 as change relative to flibanserin administered alone (test/reference).
Moderate CYP3A4/Moderate CYP2C9/Strong CYP2C19 Inhibitor (Fluconazole):
In a study of 15 healthy female subjects, a fluconazole 400 mg loading dose followed by 200 mg administered once daily for 5 days increased flibanserin 100 mg single dose exposure (AUC0-inf) 7-fold and Cmax 2.2-fold compared to flibanserin 100 mg alone. Three of 15 subjects (20%) experienced hypotension or syncope from concomitant use of fluconazole and flibanserin; therefore, the study was stopped early.
Strong CYP3A4 Inhibitor (Ketoconazole):
In a study of 24 healthy female subjects, ketoconazole 400 mg administered once daily for 5 days following a light breakfast increased flibanserin 50 mg single-dose exposure (AUC0-inf) 4.5-fold and Cmax 1.8-fold compared to flibanserin 50 mg alone.
Strong CYP3A4 Inhibitor (Itraconazole):
In a study of 12 healthy male and female subjects, itraconazole 200 mg administered once daily for 4 days following a loading dose of 400 mg increased flibanserin 50 mg single dose exposure (AUC0-inf) 2.6-fold and Cmax 1.7-fold when flibanserin was given 2 hours after itraconazole on Day 5, compared to exposures with flibanserin 50 mg alone. The 200 mg itraconazole dose does not maximally inhibit the CYP3A4 enzyme.
Moderate CYP3A4 Inhibitor (Grapefruit Juice):
In a study of 26 healthy female subjects, grapefruit juice (240 mL) increased flibanserin 100 mg single dose exposure (AUC0-inf) by 1.4-fold and Cmax 1.1-fold compared to flibanserin 100 mg alone.
Weak CYP3A4 Inhibitor (Oral Contraceptives):
In a meta-analysis of 17 oral contraceptive users and 91 non-users in Phase 1 studies, the oral contraceptive users had a 1.4-fold higher flibanserin AUC and 1.3‑fold higher Cmax compared to the non-users.
Strong CYP2D6 Inhibitor (Paroxetine):
Paroxetine is a strong CYP2D6 inhibitor. In a study of 19 healthy male and female subjects, flibanserin exposure decreased by approximately 4% when flibanserin 50 mg twice daily was given with paroxetine compared to flibanserin alone. Paroxetine was dosed at 20 mg once daily for 3 days followed by 40 mg once daily for 7 days.
Drugs that Decrease Flibanserin Exposure
Strong CYP3A4 Inducer (Rifampin):
In a study of 24 healthy female subjects, rifampin 600 mg given once daily for 7 days prior to administration of 100 mg flibanserin significantly decreased flibanserin exposure by 95%.
Moderate CYP3A4 Inducer (Etravirine):
Steady state etravirine, a moderate CYP3A4 inducer, decreased flibanserin exposures by approximately 21%.
Table 1. Drugs That Increase Flibanserin Exposure:
| Coadministered Drug(s) and Dose(s) | Dose of Flibanserin | n | Geometric Mean Ratio (90% Confidence Interval) of Pharmacokinetic Parameters of Flibanserin with/without Coadministered Drug No Effect=1.00 | |
|---|---|---|---|---|
| Cmax | AUC0-inf | |||
| Fluconazole 200 mg | 100 mg | 15 | 2.2 (1.8–2.8) | 7.0 (6.0–8.2) |
| Ketoconazole 400 mg | 50 mg | 24 | 1.8 (1.7–2.1) | 4.5 (4.0–5.1) |
| Itraconazole 200 mg* | 50 mg | 12 | 1.7 (1.4–2.0) | 2.6 (2.1–3.0) |
| Oral Contraceptives | 50 mg | 39 | 1.3 (1.1–1.6) | 1.4 (1.2–1.7) |
| Paroxetine 40 mg | 50 mg twice daily | 19 | 1.0 (0.9–1.2) | 1.0 (0.9–1.0) |
* itraconazole dose was not optimal for maximal inhibition of CYP3A4 enzyme.
Effects of Flibanserin on Other Drugs
The effects of flibanserin on the pharmacokinetics of other drugs are presented in Table 2 as change relative to the other drug administered alone (test/reference).
Digoxin and P-glycoprotein Substrates:
A single center, open-label, randomized, two-way crossover study in 24 healthy men and women evaluated the effect of flibanserin on the pharmacokinetics of digoxin. Flibanserin 100 mg was administered once daily over 5 days followed by a single dose of 0.5 mg digoxin, a P-gp substrate. Flibanserin increased digoxin AUC0-inf by 2.0-fold and Cmax by 1.5-fold, compared to digoxin alone.
Drugs Metabolized by CYP3A4 (Simvastatin):
An open-label, randomized, crossover study in 12 healthy men and women evaluated the effect of flibanserin 50 mg twice daily for 4 days on the pharmacokinetics of simvastatin 40 mg once daily. Flibanserin increased the AUC0-inf of simvastatin, a substrate of CYP3A4, 1.3‑fold and Cmax by 1.2-fold. Flibanserin co-administered with simvastatin increased simvastatin acid AUC0-inf by 1.5-fold and Cmax by 1.4-fold.
Oral Contraceptives:
A study in 24 healthy women evaluated the effect of 100 mg flibanserin once daily for 2 weeks on the pharmacokinetics of a single-dose of ethinyl estradiol (EE) 30 mcg/levonorgestrel (LNG) 150 mcg. Flibanserin increased the EE AUC0-inf by 1.09-fold and the EE Cmax by 1.1-fold. Flibanserin decreased the LNG AUC0-inf by 1.06-fold and did not change the LNG Cmax.
Drugs Metabolized by CYP2B6 (Bupropion):
An open-label, randomized, two-period crossover study in 28 healthy women evaluated the effect of flibanserin on the pharmacokinetics of bupropion. Flibanserin 50 mg twice daily was administered for 2 days followed by 100 mg once daily for 13 days. Bupropion 150 mg twice daily was given for 8 days beginning on Day 6 of flibanserin treatment. Flibanserin did not change bupropion AUCt,ss (1.0-fold change) and Cmax (1.0-fold change) but hydroxybupropion AUCt,ss decreased by 9% and Cmax by 11%.
Table 2. Effects of Flibanserin on Exposure of Other Drugs:
| Coadministered Drug(s) and Dose(s) | Dose of Flibanserin | n | Geometric Mean Ratio (90% Confidence Interval) of Pharmacokinetic Parameters of Coadministered Drug with/without Flibanserin No Effect=1.00 | |
|---|---|---|---|---|
| Cmax | AUC0-inf | |||
| Simvastatin 40 mg | 50 mg twice daily | 12 | 1.7 (1.4–2.0) | 2.6 (2.1–3.1) |
| Digoxin 0.5 mg | 100 mg | 24 | 1.5 (1.3–1.6) | 2.0 (1.5–2.5) |
| Ethinyl estradiol 30 mcg/ Levonorgestrel 150 mcg | 100 mg | 24 | 1.1 (1.0–1.1) 1.0 (0.9–1.0) | 1.1 (1.0–1.2) 1.0 (0.9–1.1) |
| Bupropion 150 mg | 100 mg | 28 | 1.0 (0.9–1.1) | 1.0 (1.0–1.1) |
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.