Gepirone
Chemical formula: C₁₉H₂₉N₅O₂ Molecular mass: 359.474 g/mol PubChem compound: 55191
Mechanism of action
The mechanism of the antidepressant effect of gepirone is not fully understood but is thought to be related to its modulation of serotonergic activity in the CNS through selective agonist activity at 5HT1A receptors.
Pharmacodynamic properties
The pharmacological activity of gepirone is attributed to the parent drug and its major metabolites 3'-OH-gepirone and 1-PP. Gepirone and its 3'-OH metabolite bind to 5HT1A receptors (Ki = 38 nM and 58 nM, respectively), where they act as agonists, while the 1-PP metabolite binds to alpha2 receptors (Ki = 42 nM).
Cardiac Electrophysiology
In a thorough QT study, the largest mean increase in baseline- and placebo-corrected QTc interval with administration of 100 mg per day immediate-release formulation of gepirone was 18.4 msec (upper 90% confidence interval [CI] = 22.7 ms) on Day 1 and 16.1 msec (upper 90% CI = 20.7 ms) on Day 7. The exposure in this study was 2-fold the exposure of the maximum recommended dose.
Pharmacokinetic properties
The pharmacokinetics of gepirone are linear and dose proportional from 18.2 mg to 72.6 mg. Steady-state plasma concentration are typically achieved within two to four days of daily dosing.
Absorption
The absolute bioavailability is 14% to 17%. The maximal plasma gepirone concentration (Cmax) after dosing is reached within 6 hours post dose (Tmax).
Effect of Food
After a high fat meal, Tmax is reached at 3 hours. A significant effect of food has been observed on the peak plasma concentration (Cmax) of gepirone and, to a lesser extent, on the total exposure (AUC0-tlast, AUC0-∞) to gepirone. The magnitude of the food-effect was dependent of the fat content of the meal. The systemic exposure of gepirone and major metabolites was consistently higher under fed conditions as compared to the fasted state. Gepirone Cmax after intake of low-fat (~200 calories) breakfast was 27% higher, after medium-fat (~500 calories) breakfast 55% higher and after a high-fat (~850 calories) breakfast 62% higher as compared to the fasted state. The AUC after intake of low-fat breakfast was about 14% higher, after a medium-fat breakfast 22% higher and after a high-fat breakfast 32 to 37% higher as compared to the fasted state. The effect of varying amounts of fat on Cmax and AUC of the major metabolites 3-OH-gepirone and 1PP were similar to that found for gepirone.
Distribution
The apparent volume of distribution of gepirone is approximately 94.5L. The in vitro plasma protein binding in human is 72% and is not concentration dependent. The in vitro plasma protein binding for metabolite 3'-OH gepirone is 59% and 42% for 1-PP.
Elimination
The mean terminal half-life is approximately 5 hours.
Metabolism
Gepirone is extensively metabolized and both major metabolites 1-PP and 3'-OH-gepirone are present in plasma in higher concentrations than the parent compound. CYP3A4 is the primary enzyme catalyzing the metabolism of gepirone to its major pharmacologically active metabolites.
Excretion
Following a single oral dose of [14C]-labeled gepirone, approximately 81% and 13% of the administered radioactivity was recovered in the urine and feces, respectively as metabolites. 60% of the gepirone was eliminated in the urine within the first 24 hours. The presence of hepatic or renal impairment did affect the apparent clearance of gepirone.
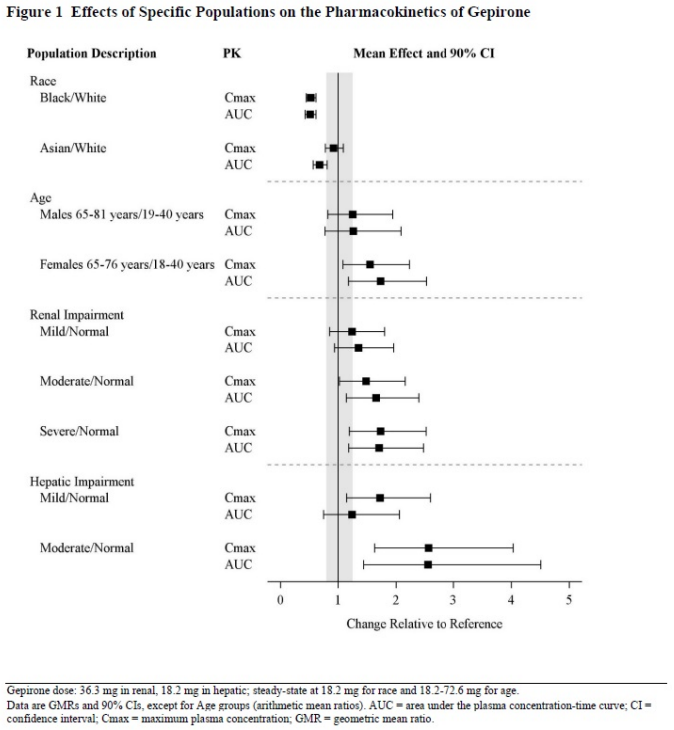
Specific Populations
Exposures of gepirone in specific populations are summarized in Figure 1.
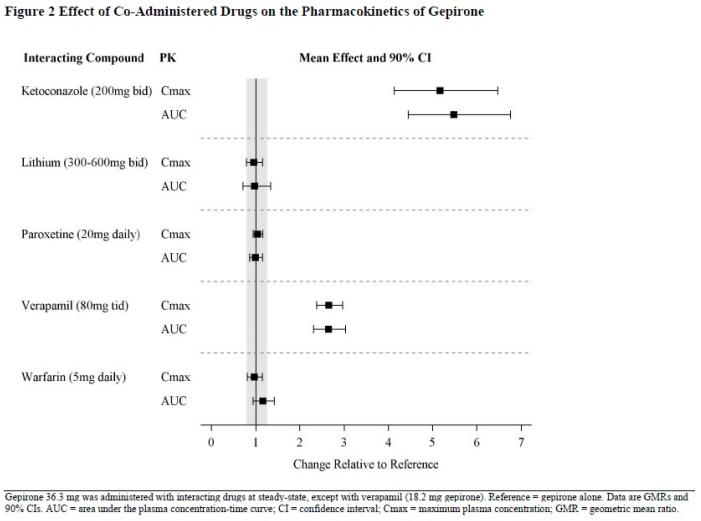
Drug Interactions Studies
In Vivo Studies
The effect of co-administered drugs on the pharmacokinetics of gepirone is summarized in Figure 2.
Rifampin:
The effect of multiple oral dosing of potent cytochrome P450 3A4 inducer rifampin on the steady-state pharmacokinetics of gepirone and its major metabolites 1-PP and 3'-OH-gepirone was investigated in 24 subjects. Combined therapy with rifampin (600 mg daily) and gepirone (20 mg for two days, then 40 mg daily) decreased Cmax and AUC0-24 of gepirone 20 times (gepirone Cmax alone 9.63 ng/mL, with rifampin 0.491 ng/mL) and 29 times (gepirone AUC0-24 alone 123 ng·hr/mL, with rifampin 4.19 ng·hr/mL), respectively. The Cmax and AUC0-24 of 3'-OH gepirone were decreased 2.5 times (3'-OH gepirone Cmax alone 23.0 ng/mL, with rifampin 9.30 ng/mL) and three times (3'-OH gepirone AUC0-24 alone 371 ng·hr/mL, with rifampin 126 ng·hr/mL), respectively. There was no effect on the pharmacokinetics of 1-PP (1-PP Cmax alone 6.37 ng/mL, with rifampin 6.02 ng/mL; 1-PP AUC0-24 alone 92.5 ng·hr/mL, with rifampin 81.1 ng·hr/mL).
Glyburide:
Under steady-state conditions for glyburide, the addition of 36.3 mg daily of gepirone for six days in 16 patients with stable Type II diabetes resulted in statistically significantly lower AUC (glyburide AUC0-12 alone 574.8 ng·h/mL, with gepirone 483.0 ng·h/mL) and Cmax (glyburide Cmax alone 121.0 ng/mL, with gepirone 96.6 ng/mL) values for glyburide.
Drugs that Interfere with Hemostasis:
Following coadministration of stable dose of warfarin (INR 1.4 to 2.0) with multiple daily doses of gepirone, no significant effect was observed in INR, prothrombin values, or total warfarin (protein bound plus free drug) pharmacokinetics for warfarin.
Drugs that Interfere with Protein Binding:
Gepirone is not highly bound to plasma protein and is not likely to be involved in interactions due to altered protein binding. In a clinical study with coadministration of gepirone (18.2 mg) and warfarin, a highly protein-bound drug, no significant change in international normalized ratio (INR) was observed.
In Vitro Studies
Gepirone at concentrations of 0.5, 5, and 50 ng/mL was shown to have no significant impact on the plasma protein binding of chlorpromazine, desipramine, diazepam, phenytoin, prazosin, propranolol, verapamil, or warfarin. The binding of digoxin and haloperidol were decreased (at maximum) by 5% and 9%, respectively. The plasma protein binding of lidocaine appeared to be increased by 4.9% in the presence of gepirone.
Alcohol:
An in vitro study showed dissolution rate for both 18.2 mg and 72.6 mg gepirone ER tablets decreased slightly as ethanol concentration increased in 0.01N HCl and 0.1N HCl at 0%, 5%, 10%, 20% and 40% alcohol. At 20 hours and 40% alcohol, approximately (mean) 76.8% and 80.7% were dissolved for the 18.2 mg and 72.6 mg gepirone ER tablets, respectively.
Transporter Systems:
Gepirone and its metabolites are unlikely to cause clinically significant inhibition of the following transporters based on in vitro data: P-gp, BCRP, BSEP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K. As such, no clinically relevant interactions with drugs metabolized/transported by these CYP enzymes or transporters would be expected.
Enzyme systems:
In addition, gepirone has not been shown to be an inhibitor or inducer of any of the cytochrome P450 enzymes. Chronic administration of gepirone is unlikely to induce the metabolism of drugs metabolized by these CYP isoforms.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.