Omadacycline
Chemical formula: C₂₉H₄₀N₄O₇ Molecular mass: 556.66 g/mol PubChem compound: 54697325
Mechanism of action
Omadacycline is an antibacterial drug.
Pharmacodynamic properties
Cardiac Electrophysiology
Based on the nonclinical and clinical data, including electrocardiogram evaluation in the phase 3 clinical trials, one of which had moxifloxacin as a control group, no clinically relevant QTc prolongation was observed at the maximum recommended dose of omadacycline.
Cardiac Physiology-Increase in Heart Rate
In phase 1 studies conducted in healthy volunteers, transient dose-dependent increases in heart rate have been observed following administration of single and multiple doses of omadacycline. The clinical implication of this finding is unknown.
In a standard radiolabeled ligand binding assays, omadacycline was shown to inhibit binding of H-scopolamine to the M2 subtype of the muscarinic acetylcholine receptor. In the heart, muscarinic M2 receptors serve as mediators of the parasympathetic input that normally is received via the vagus nerve and stimulation of the receptor increases membrane potassium conductance through the acetylcholine-dependent channel, which slows depolarization and reduces pacemaker activity in the sinoatrial node.
Pharmacokinetic properties
The pharmacokinetic parameters of omadacycline after single and multiple oral and intravenous doses are summarized in the following table.
Pharmacokinetic (PK) Parameters of Omadacycline in Healthy Adult Subjects:
| Dose and Route of Administration | 100 mg IV | 300 mg Oral | 450 mg Oral | |
|---|---|---|---|---|
| PK Parameters* | ||||
| Cmax (ng/mL) | Single dose | 1507 (38.6) | 548 (26.7) | 874 (26.6) |
| Steady state | 2120 (32.0) | 952 (44.2) | 1077 (25.0) | |
| AUC (h*ng/mL) | Single dose | 9358 (22.1) | 9399 (27.2) | 8977 (26.6) |
| Steady state | 12,140 (26.6) | 11,156 (44.9) | 13,367 (26.0) | |
| Dose Proportionality | Dose proportional increases in omadacycline Cmax and AUC following single oral doses of omadacycline from 300 to 450 mg. | |||
| Accumulation | Accumulation ratio 1.5 | |||
| Absorption | ||||
| Bioavailability | 34.5% following single 300 mg dose of omadacycline | |||
| Tmax Median (min, max) | Single dose | 0.55 (0.25, 0.68) | 2.50 (1, 4.05) | 2.50 (1.5, 3) |
| Steady state | 0.50 (0, 1) | 2.50 (0, 8) | 2.50 (1.5, 4) | |
| Distribution | ||||
| Plasma Protein Binding | 20%; not concentration dependent | |||
| Volume of Distribution (L) | Single dose | 256 (25.6) | 794 (23.6)† | ND |
| Steady state | 190 (27.7) | ND | ND | |
| Elimination | ||||
| Elimination Half-Life (hr) | Single dose | 16.2 (14.7) | 14.96 (16.5) | 13.45 (12.9) |
| Steady state | 16.0 (21.7) | 15.5 (10.7) | 16.83 (8.1) | |
| Systemic Clearance (L/hr) | Single dose | 11.24 (23.8) | 34.6 (30.9)† | ND |
| Steady state | 8.8 (25.2) | ND | ND | |
| Renal Clearance (L/hr) | 2.4 to 3.3 | |||
| Metabolism | Omadacycline is not metabolized | |||
| Excretion (Mean (SD) % dose) | Urine | 27 (3.5) % | 14.4 (2.3) %‡ | ND |
| Feces | ND | 81.1 (2.3) %‡ | ND | |
Cmax = maximum plasma concentration, AUC = area under concentration-time curve, IV = intravenous, ND = not determined, Tmax = time to Cmax
* All PK parameters presented as mean (% coefficient of variation; %CV) unless otherwise specified
† Presented as apparent clearance or volume of distribution
‡ Following administration of radiolabeled omadacycline
Absorption
The exposure to omadacycline is similar between a 300-mg oral dose and a 100-mg intravenous dose of omadacycline in healthy fasted subjects.
Effect of Food
Ingestion of a standard high-fat nondairy meal (855 calories; 59% calories from fat) and standard high-fat meal including dairy (985 calories; 60% calories from fat) 2-hours before administration of a single 300-mg oral dose of omadacycline decreased the rate (Cmax) and extent of absorption (AUC) by 40% and 42%, and 59% and 63%, respectively compared to administration of omadacycline under fasting conditions. The rate and extent of absorption of omadacycline were not substantially decreased when a high-fat nondairy meal (800-1000 calories; 50% calories from fat) was ingested 4 hours pre-dose.
Following ingestion of either a light non-fat (300-350 calories; ≤5% calories from fat), or a standard low-fat (800-1000 calories; 30% calories from fat), or a standard high fat (800-1000 calories; 50% calories from fat) meal 2 hours post-dose, the AUC and Cmax were not substantially altered, as compared to fasting conditions.
Distribution
Plasma protein binding of omadacycline is approximately 20% and is not concentration dependent. The mean (% CV) volume of distribution of omadacycline at steady-state following IV administration of omadacycline in healthy subjects was 190 (27.7) L.
Elimination
Renal clearance of omadacycline following IV administration of omadacycline ranged from 2.4 to 3.3 L/h in healthy subjects.
Metabolism
In vitro studies using human liver microsomes and hepatocytes demonstrated that omadacycline is not metabolized.
Excretion
Following a 100-mg IV dose of omadacycline, 27% of the dose was recovered as unchanged omadacycline in the urine. In healthy male volunteers receiving 300-mg oral [14C] omadacycline, 77.5% to 84.0% of the dose was recovered in the feces, approximately 14.4% (range 10.8% to 17.4%) in the urine, with 95.5% of the administered radioactive dose recovered after 7 days.
Lung Penetration
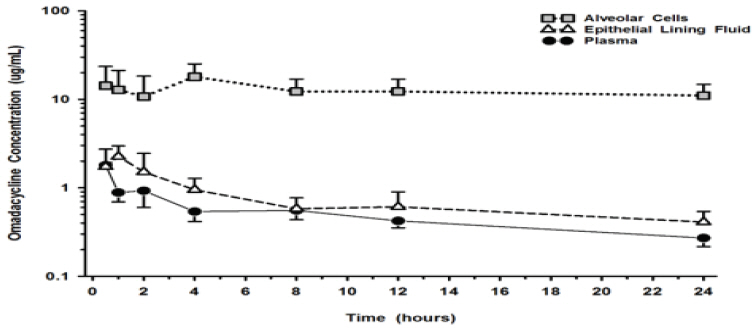
The mean omadacycline concentrations over time for alveolar cells (AC), epithelial lining fluid (ELF), and plasma following IV administration of multiple doses of 100-mg of omadacycline to healthy volunteers are shown in Figure 1. The steady-state omadacycline AUC0-24h (302.5 hr*mcg/mL) in AC was 25.8-fold higher than the plasma AUC0-24h, and the AUC0-24h (17.2 hr*mcg/mL) in ELF was 1.5-fold higher than the AUC0-24h in plasma.
Figure 1. Mean (± SD) Concentrations of Omadacycline in Alveolar Cells, Epithelial Lining, and Plasma Following Multiple 100 mg IV Doses of omadacycline to Healthy Subjects During Bronchoscopy Sampling Times:
Specific Populations
No clinically significant differences in the pharmacokinetics of omadacycline were observed based on age, gender, race, weight, renal impairment or end-stage renal disease, and hepatic impairment.
Patients with Renal Impairment
A study was conducted to compare omadacycline pharmacokinetics following 100-mg IV administration in 8 subjects with end-stage renal disease (ESRD) on stable hemodialysis, with and 8-matched healthy control subjects. In the ESRD subjects, omadacycline was administered on two separate occasions; immediately prior to dialysis and after dialysis, and the AUC, Cmax, and CL of omadacycline were comparable between the renally impaired subjects and the matching healthy subjects. During dialysis, 7.9% of omadacycline was recovered in the dialysate. Renal impairment did not impact omadacycline elimination.
Patients with Hepatic Impairment
A study was conducted to compare omadacycline pharmacokinetics following intravenous and oral dosing to 5 subjects with mild hepatic impairment (Child-Pugh Class A), 6 subjects with moderate hepatic impairment (Child-Pugh Class B), and 6 subjects with severe hepatic impairment (Child-Pugh Class C) as compared to 12 matched healthy control subjects. The AUC and Cmax of omadacycline were comparable between the hepatically impaired subjects and the matching healthy subjects, and similar clearance was observed across all cohorts. Hepatic impairment did not impact omadacycline elimination.
Drug Interaction Studies
Clinical Studies
Administration of oral verapamil (P-gp inhibitor) two hours prior to a single 300 mg oral dose of omadacycline increased omadacycline AUC by approximately 25% and Cmax by approximately 9%.
In vitro Studies
In vitro studies in human liver microsomes indicate that omadacycline does not inhibit nor induce metabolism mediated by CYP 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5, or UGT1A1. Therefore, omadacycline is not expected to alter the pharmacokinetics of drugs metabolized by the above stated human hepatic enzymes.
Omadacycline is not an inhibitor of P-gp and organic anion transporting polypeptide (OATP) 1B1 and OATP1B3. Omadacycline is a substrate of P-gp (see Clinical Studies above). Omadacycline is not a substrate or inhibitor of the major organic anion transporters (OAT-1 and 3), breast cancer resistance protein (BCRP), or multidrug resistance-associated protein 2 (MRP2). Omadacycline was not an OATP1B1 or OATP1B3 substrate at supra-therapeutic concentrations (5-13 fold higher than clinically relevant concentrations).
Preclinical safety data
Hyperpigmentation of the thyroid has been produced by members of the tetracycline class in the following species: in rats by omadacycline, oxytetracycline, doxycycline, tetracycline PO4, and methacycline; in minipigs by doxycycline, minocycline, tetracycline PO4, and methacycline; in dogs by doxycycline and minocycline; in monkeys by omadacycline and minocycline.
Minocycline, tetracycline PO4, methacycline, doxycycline, tetracycline base, oxytetracycline HCl, and tetracycline HCl were goitrogenic in rats fed a low iodine diet. This goitrogenic effect was accompanied by high radioactive iodine uptake. Administration of minocycline also produced a large goiter with high radioiodine uptake in rats fed a relatively high iodine diet.
Treatment of various animal species with this class of drugs has also resulted in the induction of thyroid hyperplasia in the following: in rats and dogs (minocycline); in chickens (chlortetracycline); and in rats and mice (oxytetracycline). Adrenal gland hyperplasia has been observed in goats and rats treated with oxytetracycline.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.