Palopegteriparatide
Pharmacodynamic properties
Endogenous parathyroid hormone (PTH) is secreted by the parathyroid glands as a polypeptide of 84 amino acids. PTH exerts its action via cell-surface parathyroid hormone receptors, for example, expressed in bone, kidney and nerve tissue. Activation of PTH1R stimulates bone turnover, increases renal calcium reabsorption and phosphate excretion and facilitates synthesis of active vitamin D.
Palopegteriparatide is a prodrug, consisting of PTH(1-34) conjugated to a methoxypolyethylene glycol carrier (mPEG) via a proprietary TransCon Linker. PTH(1-34) and its main metabolite, PTH(1-33), have similar affinity to and activation of PTH1R as endogenous PTH. At physiological conditions, PTH is cleaved from palopegteriparatide in a controlled manner to provide a continuous systemic exposure of active PTH.
Pharmacokinetic properties
Absorption
Following daily subcutaneous administration, palopegteriparatide releases PTH via autocleavage of the TransCon Linker with first-order kinetics, resulting in continuous exposure over 24 hours within the estimated normal range (figure 1).
Figure 1. Mean released PTH* following subcutaneous administration of palopegteriparatide at steady state in patients with hypoparathyroidism:
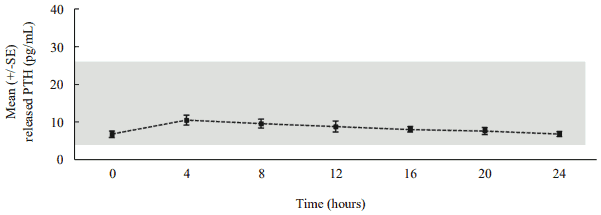
The estimated normal range for PTH(1-34) is approximately 4 to 26 pg/mL. This is calculated based on PTH(1-34) constituting 40% of the molecular weight of PTH(1-84)** and the normal range (10 to 65 pg/mL) for PTH(1-84).
* Mean palopegteriparatide dose (range): 22.3 (12-33) mcg PTH(1-34)/day, n=7, released PTH: sum of PTH(1-34) and PTH(1-33).
** PTH(1-84) = endogenous parathyroid hormone.
In patients with hypoparathyroidism administered palopegteriparatide corresponding to 18 mcg of PTH(1-34)/day, the predicted maximum plasma concentration (Cmax) (CV%) of palopegteriparatide was 5.18 ng/mL (36%) and the predicted Cmax (CV%) for released PTH was 6.9 pg/mL (22%) with a median time to reach maximum concentrations (Tmax) of 4 hours. The predicted exposure over the 24-hour dosing interval (area under the curve, AUC) (CV%) for released PTH was 150 pg*h/mL (22%).
Following multiple subcutaneous doses of palopegteriparatide in the range of 12 to 24 mcg PTH(1-34)/day, the palopegteriparatide and released PTH concentrations increased in a dose-proportional manner reaching steady-state within approximately 10 and 7 days, respectively. The peak-to-trough ratio was low, approximately 1.1 and 1.5 over 24 hours at steady state for palopegteriparatide and released PTH, respectively. Palopegteriparatide accumulated after multiple dosing by up to 18-fold for AUC.
Distribution
The apparent volume of distribution (CV%) of palopegteriparatide is estimated to 4.8 L (50%) and to 8.7 L (18%) for released PTH.
Biotransformation
PTH released from palopegteriparatide is composed of PTH(1-34) and the metabolite PTH(1-33). PTH is renally metabolised and cleared.
Elimination
In healthy adults, the clearance (CV%) of palopegteriparatide at steady state is estimated to be 0.58 L/day (52%) with a predicted half-life of 70 hours. The apparent half-life of PTH released from palopegteriparatide is approximately 60 hours. In the liver, most of the PTH is cleaved by cathepsins. In the kidney, a small amount of PTH binds to PTH1R, but most is excreted by glomerular filtration.
Pharmacokinetic/pharmacodynamic relationship
In a pharmacodynamic/pharmacokinetic sub-study in hypoparathyroid patients, daily subcutaneous administration of palopegteriparatide (mean dose (range): 22.3 (12-33) mcg PTH(1-34)/day) increased serum calcium levels to within the normal range (see figure 2). The increase in serum calcium levels occurred in a dose-related manner, supporting the ability to titrate palopegteriparatide according to measured serum calcium values in the individual patient.
Figure 2. Mean albumin-adjusted serum calcium concentrations following subcutaneous administration of palopegteriparatide at steady state in patients with hypoparathyroidism:
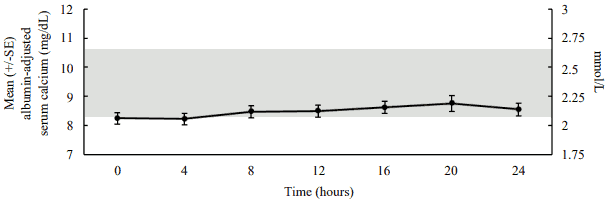
The normal range for albumin-adjusted serum calcium is 2.07 to 2.64 mmol/L (8.3 to 10.6 mg/dL) as denoted by the grey shading. Mean palopegteriparatide dose (range): 22.3 (12-33) mcg PTH(1-34)/day, n=7.
Special populations
The pharmacokinetics of released PTH was not influenced by sex or body weight. The data for race and ethnicity did not show any trends indicating differences, but the available data are too limited to make definitive conclusions.
Elderly
The pharmacokinetics of released PTH was not influenced by age (19 to 76 years old).
Renal impairment
Palopegteriparatide has been administered to patients with hypoparathyroidism with an eGFR of ≥30 mL/min in long-term clinical trials without the need for dose adjustment beyond the trial titration algorithm. No clinical trials were conducted in patients with hypoparathyroidism with severe renal impairment (<30 mL/min) or on dialysis. In a trial where palopegteriparatide was administered as a single dose to non-hypoparathyroid subjects with renal impairment, palopegteriparatide exposure and resulting serum calcium levels were similar in subjects with mild, moderate, and severe renal impairment as compared to subjects without renal impairment.
Preclinical safety data
No special hazard for humans were revealed in the conventional studies of safety pharmacology, genotoxicity, and local tolerance conducted with palopegteriparatide.
At the highest dose levels in all animal species employed, repeated dosing resulted in adverse persistent hypercalcemia, which in some studies led to premature death/euthanasia, clinical signs, body weight loss and/or soft tissue mineralisation observed mainly in the kidneys. These findings are considered results of persistent exaggerated PTH pharmacology and of no relevance in a clinical setting where dose adjustments are performed to ensure normalised serum calcium.
In accordance with the expected pharmacological effects, repeated daily administration of palopegteriparatide increased bone turnover in rats. At low dose levels (2-fold the maximum recommended human dose (MRHD), based on exposure to released PTH by AUC) in rats, the increased bone turnover induced overall net catabolic bone effects. At high dose levels (5-fold the MRHD, based on exposure to released PTH by AUC) in rats, the increased bone turnover resulted in a net anabolic bone effect. Physeal dysplasia was observed at the highest dose level (9-fold the MRHD, based on exposure to released PTH by AUC) in rats. These effects are of no relevance in a clinical setting where palopegteriparatide doses are individually adjusted.
There were no cardiovascular findings in monkeys up to and including the highest dose tested in single- (3-fold the MRHD, based on exposure to released PTH by Cmax) or repeat-dose studies (0.98-fold the MRHD, based on exposure to released PTH by Cmax).
Increased occurrence of osteosarcomas has been observed in carcinogenicity studies with short-lived PTH analogues in rats, but there is no evidence of increased risk of osteosarcoma in patients treated with short-lived PTH analogues. No carcinogenicity study has been conducted with palopegteriparatide.
In animal reproduction studies, administration of palopegteriparatide to pregnant rats and rabbits during the period of organogenesis resulted in no evidence of embryo-lethality, foetotoxicity or dysmorphogenesis up to and including the highest doses tested (8- and 7-fold, respectively, the MRHD, based on exposure to released PTH by AUC). Exaggerated PTH pharmacological effects were observed at the highest doses tested in the pregnant rats and rabbits (increased serum calcium levels, decreased body weight, decreased food consumption and/or clinical signs). The exposures at the no observed adverse effect level (NOAEL) for maternal toxicity were 2- and 3-fold the MRHD, based on exposure to released PTH by AUC in pregnant rats and rabbits, respectively. A pre- and postnatal developmental study has not been conducted with palopegteriparatide.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.