Sotagliflozin
Chemical formula: C₂₁H₂₅ClO₅S Molecular mass: 424.94 g/mol PubChem compound: 24831714
Mechanism of action
Sotagliflozin is an inhibitor of SGLT2 and SGLT1. Inhibiting SGLT2 reduces renal reabsorption of glucose and sodium which may influence several physiological functions such as lowering both pre-and afterload of the heart and downregulating sympathetic activity. Inhibiting SGLT1 reduces intestinal absorption of glucose and sodium which likely contributes to diarrhea. The mechanism for sotagliflozin's cardiovascular benefits has not been established.
Pharmacodynamic properties
Cardiac Electrophysiology
In a randomized, placebo-controlled, active-comparator, crossover study, 58 healthy subjects were administered a single oral dose of sotagliflozin 800 mg or sotagliflozin 2000 mg (5 times the maximum recommended dose), moxifloxacin, and placebo. No increase in QT corrected for heart rate (QTc) was observed with either 800 mg or 2000 mg sotagliflozin.
Pharmacokinetic properties
Plasma Cmax and AUC of sotagliflozin increased in a dose-proportional manner in the therapeutic dose range of 200 mg to 400 mg once daily. Accumulation of sotagliflozin was observed with an approximate 50-100% increase in both Cmax and area under the concentration-time curve from 0 to 24 hours (AUC0-24h) at steady state verses the first day of dosing.
Absorption
The absolute bioavailability of oral sotagliflozin 400 mg dose was approximately 25% (90% confidence interval [CI] 16% to 39%) for area under the concentration-time curve from time 0 to last measurable concentration (AUC0-last). The contribution of enterohepatic circulation to the overall exposure is estimated to be approximately 50%. The median time to maximum plasma concentration (Tmax) ranged from 1.25 to 3 hours, over a single-dose range of 200 mg to 2000 mg. Following administration of multiple doses (400 mg and 800 mg), the median Tmax values ranged from 2.5 to 4 hours.
Food Effect
When sotagliflozin tablets were administered with a high-caloric breakfast compared to fasting conditions, plasma exposure to sotagliflozin as measured by Cmax and AUC0-inf increased by 149% and 50%, respectively. Multiple doses of sotagliflozin 400 mg given immediately before breakfast, 30 minutes prior to breakfast, and 1-hour before breakfast in healthy subjects showed a consistent effect of sotagliflozin on urine glucose excretion (UGE), insulin, and postprandial glucose (PPG) across all dose schedules. It is recommended that sotagliflozin be taken not more than one hour before the first meal of the day.
Distribution
Both sotagliflozin and its major human metabolite sotagliflozin 3-O-glucuronide exhibited high binding to human plasma proteins in vitro (>93% bound), which was not dependent on the concentration of sotagliflozin and sotagliflozin 3-O-glucuronide and was not influenced by the degree of renal or hepatic function.
Following administration of a single 400 mg dose of [14C]-sotagliflozin, the mean blood/plasma ratios for total radioactivity were 0.5 and 0.4 for Cmax and AUC0-last, respectively, and the mean whole blood to plasma concentration ratio of sotagliflozin ranged from 0.5 to 0.6.
The mean apparent volume of distribution of sotagliflozin following administration of a single 400 mg oral dose of [14C]-sotagliflozin was 9000 L.
Elimination
Metabolism
Following the administration of a single dose of 400 mg [14C]-sotagliflozin in healthy subjects, sotagliflozin was extensively metabolized predominantly to sotagliflozin 3-O-glucuronide and it represented 94.3% of the radioactivity in plasma.
The key enzymes responsible for the metabolism of sotagliflozin were UGT1A9 and, to a lesser extent, CYP3A4. Following incubation of sotagliflozin with UGT1A9, the 3-O-glucuronide metabolite of sotagliflozin was the main conjugate observed. No acyl glucuronides of sotagliflozin were identified.
Excretion
Following the administration of a single dose of 400 mg [14C]-sotagliflozin, 57% and 37% of the radioactivity was recovered in the urine and feces, respectively. These results indicate that the main route of elimination was renal. The predominant metabolite detected in urine was sotagliflozin 3-O-glucuronide, representing a mean of 33% of the administered radioactive dose. Unchanged [14C]-sotagliflozin was the predominant radioactive peak detected in fecal extracts representing a mean of 23% of the total administered radioactive dose. Sotagliflozin undergoes enterohepatic circulation. Following administration of 200 mg and 400 mg, mean terminal half-life (T1/2) ranged from 21 to 35 hours for sotagliflozin and from 19 to 26 hours for sotagliflozin 3-O-glucuronide. Effective half-life of sotagliflozin ranged from 5 to 10 hours. Steady state was generally achieved after 5 days of once daily dosing with sotagliflozin 200 mg and 400 mg.
In healthy volunteers, mean oral clearance (CL/F) of sotagliflozin ranged from 260 to 370 L/hr following administration of 200 mg and 400 mg sotagliflozin. The median population PK model predicted CL/F in type 2 diabetes mellitus patients with normal renal function was about 300 L/hr.
Specific Populations
Renal Impairment
Exposure of sotagliflozin was evaluated in a dedicated PK study in subjects with mild (eGFR 60 to <90 mL/min/1.73 m²) and moderate (eGFR 30 to <60 mL/min/1.73 m²) renal impairment and with normal renal function (eGFR ≥90 mL/min/1.73 m²). Exposure to sotagliflozin following a single dose of 400 mg was approximately 70% higher in subjects with mild and up to 170% higher in subjects with moderate renal impairment compared to subjects with normal renal function.
Hepatic Impairment
In a study with subjects with reduced hepatic function, AUC of sotagliflozin was not increased in mild (Child Pugh A) hepatic impaired subjects but was increased by approximately 3-fold in moderate (Child Pugh B) and approximately 6-fold in severe (Child Pugh C) hepatic impaired subjects compared to subjects with normal hepatic function.
Effects of Age, Sex, Race, and Body Weight on Pharmacokinetics
Based on population PK analysis, age, body weight, sex and race (non-white versus primarily whites) do not have a clinically meaningful effect on the PK of sotagliflozin.
Drug Interactions
In Vitro Studies
The major human metabolite of sotagliflozin, sotagliflozin 3-O-glucuronide, was shown to have an in vitro potential to inhibit CYP3A4 and CYP2D6 and to induce CYP3A4.
Sotagliflozin was shown to have inhibitory effects on P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Sotagliflozin is a substrate of the human uptake transporters organic anion transporter (OAT)3, organic-anion-transporting polypeptides (OATP)1B1, and OATP1B3, but not OAT1 and organic cation transporter 2 (OCT2). Sotagliflozin does not inhibit any of these human uptake transporters at clinically relevant plasma concentrations.
In Vivo Studies
Figure 1 summarizes the effect on sotagliflozin by concomitantly administered drugs. The observed changes in overall exposure (AUC) of sotagliflozin following coadministration with hydrochlorothiazide, ramipril, metformin, mefenamic acid, and oral contraceptives are not considered to be clinically relevant. Rifampicin (UGT inducer) decreases exposure to sotagliflozin.
Figure 1. Effect of Various Medications on the Pharmacokinetics of Sotagliflozin, Displayed as Geometric Mean AUCs and Cmax Ratios:
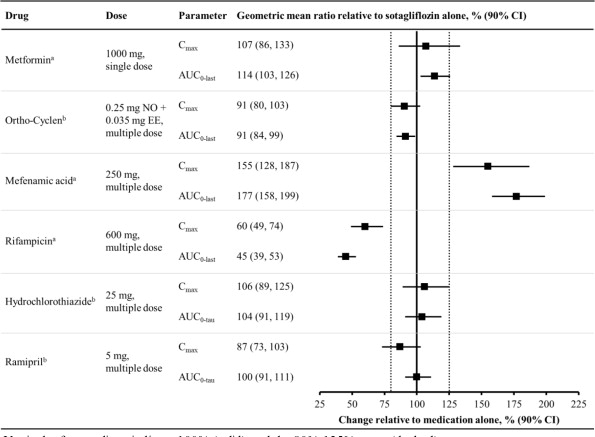
Vertical reference lines indicate 100% (solid) and the 80%-125% range (dashed)
a 400 mg sotagliflozin, administered as a single dose
b 400 mg sotagliflozin, at steady state
AUC0-last = area under the concentration-time curve from time 0 to last measurable concentration; AUC0-tau = area under the concentration-time curve from time 0 to end of the dosing period; Cmax = maximum drug concentration; EE = ethinyl estradiol; NO = norgestimate
Figure 2 summarizes the effect on concomitantly administered drugs by sotagliflozin. The increases in exposure (AUC) of metoprolol (CYP2D6 substrate) and rosuvastatin (BCRP substrate), as well as the decrease in exposure to midazolam (CYP3A4 substrate) are not considered to be clinically relevant. The increased exposure (Cmax and AUC) in ramipril is not considered clinically significant because the exposure of ramiprilat, the primary active metabolite, is minimally increased.
The increase in exposure (Cmax and AUC) of digoxin, a P-gp substrate, when coadministered with sotagliflozin, requires monitoring of patients.
Figure 2. Effect of Sotagliflozin on the Pharmacokinetics of Various Medications, Displayed as Geometric Mean AUCs and Cmax Ratios:
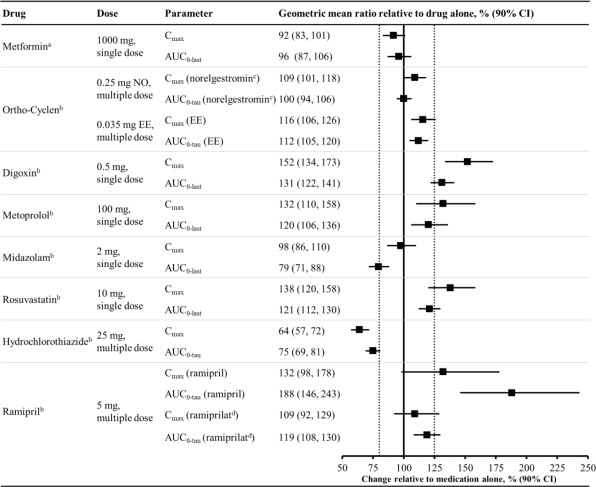
Vertical reference lines indicate 100% (solid) and the 80%-125% range (dashed)
a 400 mg sotagliflozin, administered as a single dose
b 400 mg sotagliflozin, at steady state
c Norelgestromin is the primary active metabolite of NO
d Ramiprilat is the primary active metabolite of ramipril
AUC0-last = area under the concentration-time curve from time 0 to last measurable concentration; AUC0-tau = area under the concentration-time curve from time 0 to end of the dosing period; Cmax maximum drug concentration; EE = ethinyl estradiol; NO = norgestimate
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.