Angiox 250mg powder for concentrate for solution for injection or infusion Ref.[2569] Active ingredients: Bivalirudin
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2012 Publisher: The Medicines Company UK Ltd 115L Milton Park Abingdon Oxfordshire OX14 4SA UNITED KINGDOM
Pharmacodynamic properties
Pharmacotherapeutic group: Direct thrombin inhibitors
ATC code: B01AE06
Angiox contains bivalirudin, a direct and specific thrombin inhibitor that binds both to the catalytic site and the anion-binding exosite of fluid-phase and clot-bound thrombin.
Thrombin plays a central role in the thrombotic process, acting to cleave fibrinogen into fibrin monomers and to activate Factor XIII to Factor XIIIa, allowing fibrin to develop a covalently cross-linked framework that stabilises the thrombus. Thrombin also activates Factors V and VIII, promoting further thrombin generation, and activates platelets, stimulating aggregation and granule release. Bivalirudin inhibits each of these thrombin effects.
The binding of bivalirudin to thrombin, and therefore its activity, is reversible as thrombin slowly cleaves the bivalirudin, Arg3-Pro4, bond, resulting in recovery of thrombin active site function. Thus, bivalirudin initially acts as a complete non-competitive inhibitor of thrombin, but transitions over time to become a competitive inhibitor enabling initially inhibited thrombin molecules to interact with other clotting substrates and to coagulation if required.
In vitro studies have indicated that bivalirudin inhibits both soluble (free) and clot-bound thrombin. Bivalirudin remains active and is not neutralised by products of the platelet release reaction.
In vitro studies have also shown that bivalirudin prolongs the activated partial thromboplastin time (aPTT) thrombin time (TT) and pro-thrombin time (PT) of normal human plasma in a concentration-dependent manner and that bivalirudin does not induce a platelet aggregation response against sera from patients with a history of Heparin-Induced Thrombocytopenia/Thrombosis Syndrome (HIT/HITTS).
In healthy volunteers and patients, bivalirudin exhibits dose- and concentration-dependent anticoagulant activity as evidenced as prolongation of the ACT, aPTT, PT, INR and TT. Intravenous administration of bivalirudin produces measurable anticoagulation within minutes.
The pharmacodynamic effects of bivalirudin may be assessed using measures of anticoagulation including the ACT. The ACT value is positively correlated with the dose and plasma concentration of bivalirudin administered. Data from 366 patients indicates that the ACT is unaffected by concomitant treatment with a GP IIb/IIIa inhibitor.
In clinical studies bivalirudin has been shown to provide adequate anticoagulation during PCI procedures.
The HORIZONS Trial (Patients with STEMI undergoing primary PCI)
The HORIZONS trial was a prospective, dual arm, single blind, randomised, multi-centre trial to establish the safety and efficacy of bivalirudin in patients with STEMI undergoing a primary PCI strategy with stent implantation with either a slow release paclitaxal-eluding stent (TAXUS™) or an otherwise identical uncoated bare metal stent (Express2™). A total of 3,602 patients were randomised to receive either bivalirudin (1,800 patients) or unfractionated heparin plus a GP IIb/IIIa inhibitor (1,802 patients). All patients received aspirin and clopidogrel with twice as many patients (approximately 64%) receiving a 600mg loading dose of clopidogrel than a 300mg loading dose of clopidogrel. Approximately 66% of patients were pre-treated with unfractionated heparin.
The dose of bivalirudin used in HORIZONS was the same as that used in the REPLACE-2 study (0.75 mg/kg bolus followed by a 1.75 mg/kg body weight/hour infusion). A total of 92.9% of patients treated underwent primary PCI as their primary management strategy.
The analysis and results for the HORIZONS trial at 30 days for the overall (ITT) population is shown in Table 2.Results at 1 year were consistent with results at 30 days.
Bleeding definitions and outcomes from the HORIZONS trial are shown in Table 6.
Table 2 - HORIZONS 30-day study results (intent-to-treat population):
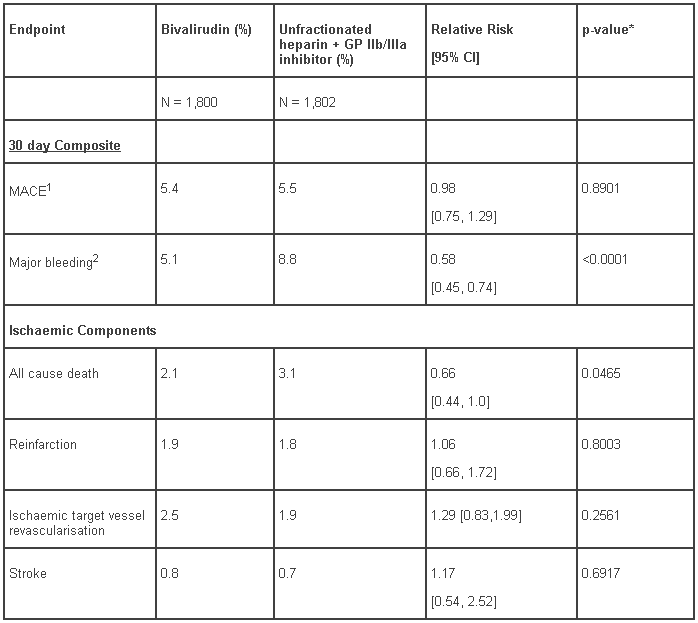
* Superiority p-value.
1 Major Adverse Cardiac/Ischaemic Events (MACE) was defined as the occurrence of any of the following; death, reinfarction, stroke or ischaemic target vessel revascularisation.
2 Major bleeding was defined using the ACUITY bleeding scale.
ACUITY Trial (Patients with unstable angina/non-ST segment elevated myocardial infarction (UA/NSTEMI)
The ACUITY trial was a prospective, randomised open-label, trial of bivalirudin with or without GP IIb/IIIa inhibitor (Arms B and C respectively) versus unfractionated heparin or enoxaparin with GP IIb/IIIa inhibitor (Arm A) in 13,819 high risk ACS patients.
In Arms B and C of the ACUITY trial, the recommended dose of bivalirudin was an initial post-randomisation IV bolus of 0.1 mg/kg followed by a continuous IV infusion of 0.25 mg/kg/h during angiography or as clinically warranted.
For patients undergoing PCI, an additional IV bolus of 0.5 mg/kg bivalirudin was administered and the rate of IV infusion increased to 1.75 mg/kg/h.
In Arm A of the ACUITY trial, UFH or enoxaparin was administered in accordance with the relevant guidelines for the management of ACS in patients with UA and NSTEMI. Patients in Arms A and B were also randomised to receive a GP IIb/IIIa inhibitor either upfront at the time of randomization (prior to angiography) or at the time of PCI. A total of 356 (7.7%) of patients randomised to Arm C also received a GP IIb/IIIa inhibitor.
High risk patient characteristics of the ACUITY population that mandated angiography within 72 hours were balanced across the three treatment arms. Approximately 77% of patients had recurrent ischaemia, approximately 70% had dynamic ECG changes or elevated cardiac biomarkers, approximately 28% had diabetes and approximately 99% of patients underwent angiography within 72 hours.
Following angiographic assessment, patients were triaged to either medical management (33%), PCI (56%) or CABG (11%). Additional anti-platelet therapy utilised in the study included aspirin and clopidogrel.
The primary analysis and results for ACUITY at 30-days and 1 year for the overall (ITT) population and for the patients that received aspirin and clopidogrel as per protocol (pre-angiography or pre-PCI) are shown in Tables 3 and 4.
Table 3 - ACUITY trial; 30-day and 1-year risk differences for the composite ischaemic endpoint and its components for the overall population (ITT):
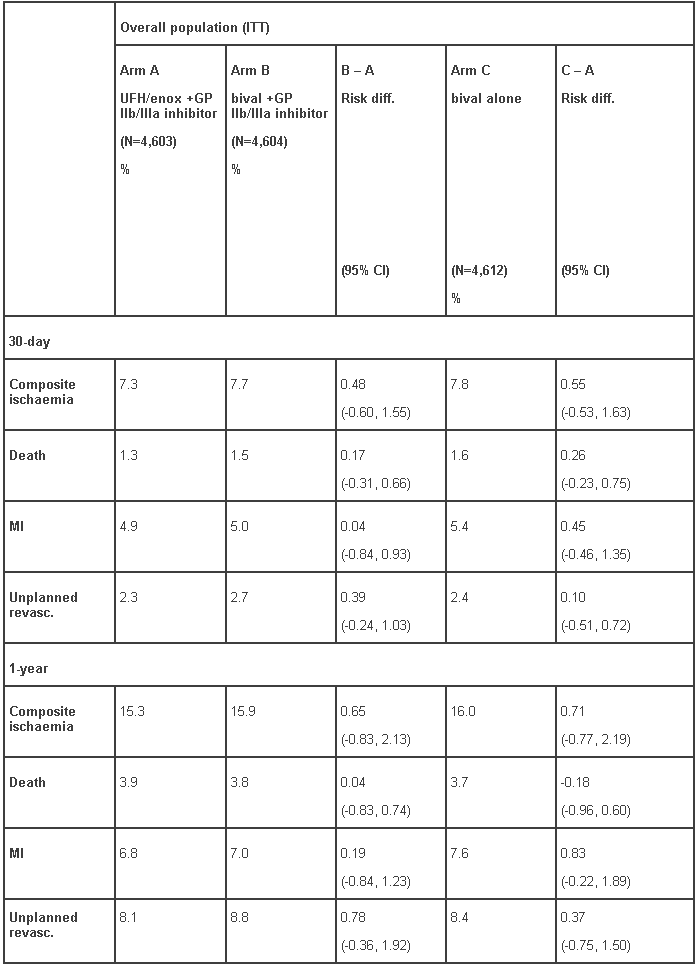
Table 4 - ACUITY trial; 30-day and 1-year risk differences for the composite ischaemic endpoint and its components for patients that received aspirin and clopidogrel as per protocol:*
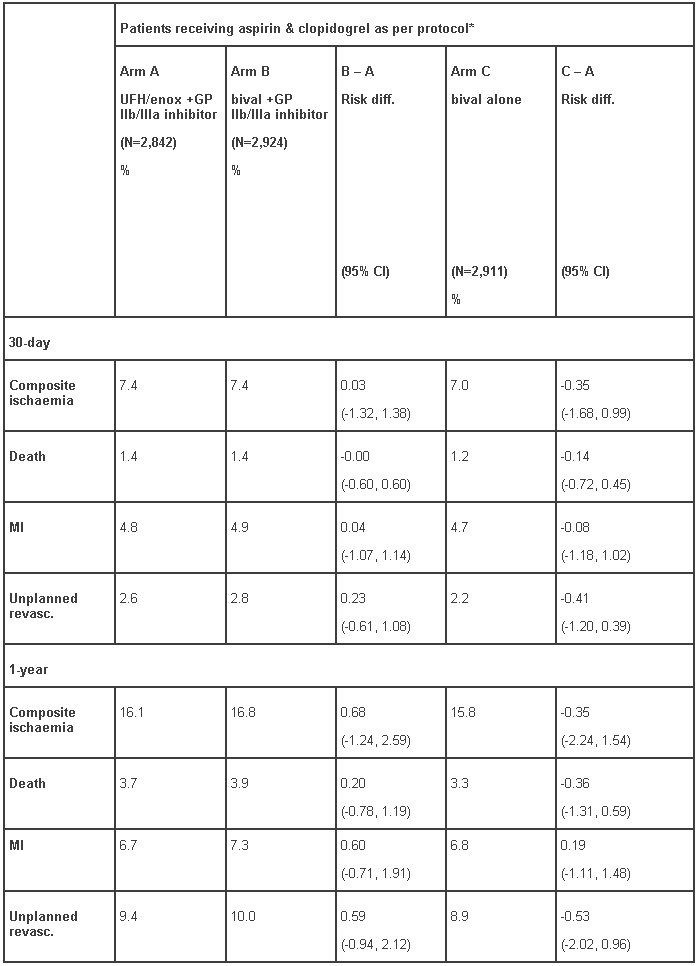
* clopidogrel pre-angiography or pre-PCI
The incidence of both ACUITY-scale and TIMI-scale bleeding events up to day 30 for the intent-to-treat population is presented in Table 6. The incidence of both ACUITY-scale and TIMI-scale bleeding events to day 30 for the per protocol population are presented in Table 7. The advantage of bivalirudin over UFH/enoxaparin plus GP IIb/IIIa inhibitor in terms of bleeding events was only observed in the bivalirudin monotherapy arm.
The REPLACE-2 Trial (Patients undergoing PCI)
The 30-day results based on quadruple and triple endpoints from a randomized, double-blind trial of over 6,000 patients undergoing PCI (REPLACE-2) are shown in Table 5. Bleeding definitions and outcomes from the REPLACE-2 trial are shown in Table 6.
Table 5 - REPLACE-2 study results: 30-day endpoints (intent-to-treat and per-protocol populations:
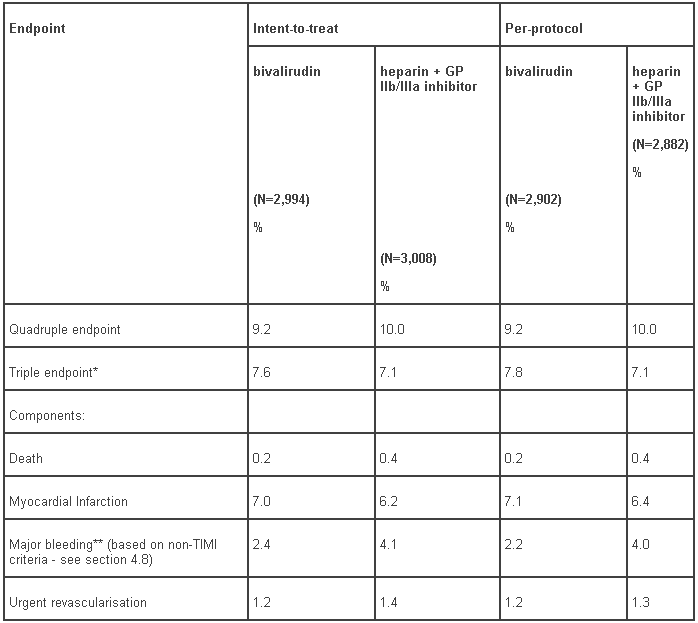
* excludes major bleeding component
** p<0.001
Table 6 - Major bleeding rates in clinical trials of bivalirudin 30 day endpoints for intent-to-treat populations:
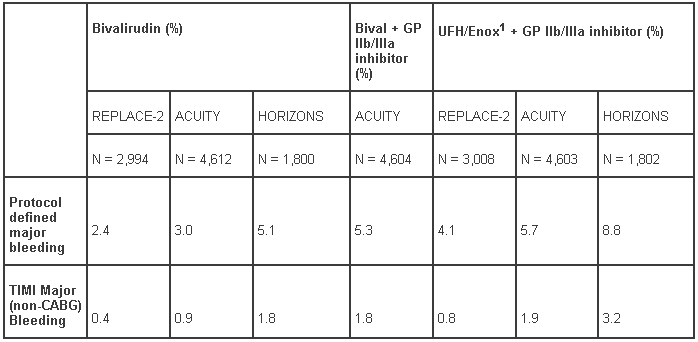
1 Enoxaparin was used as comparator in ACUITY only.
Table 7 - ACUITY trial; bleeding events up to day 30 for the population of patients who received aspirin and clopidogrel as per protocol:*

* clopidogrel pre-angiography or pre-PCI
Bleeding Definitions
REPLACE-2 major bleeding was defined as the occurrence of any of the following: intracranial haemorrhage, retroperitoneal haemorrhage, blood loss leading to a transfusion of at least two units of whole blood or packed red blood cells, or bleeding resulting in a haemoglobin drop of more than 3 g/dl, or a fall in haemoglobin greater than 4 g/dl (or 12% of haematocrit) with no bleeding site identified. ACUITY major bleeding was defined as any one of the following: intracranial, retroperitoneal, intraocular, access site haemorrhage requiring radiological or surgical intervention, ≥5 cm diameter haematoma at puncture site, reduction in haemoglobin concentration of ≥4 g/dl without an overt source of bleeding, reduction in haemaglobin concentration of ≥3 g/dl with an overt source of bleeding, re-operation for bleeding, use of any blood product transfusion. Major bleeding in the HORIZONS study was also defined using the ACUITY scale. TIMI major bleeding was defined as intracranial bleeding or a decrease in haemoglobin concentration ≥5 g/dl.
Heparin-induced thrombocytopenia (HIT) and heparin-induced thrombocytopenia-thrombosis syndrome (HIT/HITTS)
Clinical trials in a small number of patients have provided limited information about the use of Angiox in patients with HIT/HITTS.
Pharmacokinetic properties
The pharmacokinetic properties of bivalirudin have been evaluated and found to be linear in patients undergoing Percutaneous Coronary Intervention and in patients with ACS.
Absorption: The bioavailability of bivalirudin for intravenous use is complete and immediate. The mean steady-state concentration of bivalirudin following a constant intravenous infusion of 2.5 mg/kg/h is 12.4 µg/ml.
Distribution: Bivalirudin is rapidly distributed between plasma and extracellular fluid. The steady-state volume of distribution is 0.1 l/kg. Bivalirudin does not bind to plasma proteins (other than thrombin) or to red blood cells.
Biotransformation: As a peptide, bivalirudin is expected to undergo catabolism to its constituent amino acids, with subsequent recycling of the amino acid in the body pool. Bivalirudin is metabolized by proteases, including thrombin. The primary metabolite resulting from the cleavage of Arg3-Pro4 bond of the N-terminal sequence by thrombin is not active because of the loss of affinity to the catalytic active site of thrombin. About 20% of bivalirudin is excreted unchanged in the urine.
Elimination: The concentration-time profile following intravenous administration is well described by a two-compartment model. Elimination follows a first order process with a terminal half-life of 25 ± 12 minutes in patients with normal renal function. The corresponding clearance is about 3.4 ± 0.5 ml/min/kg.
Hepatic Insufficiency: The pharmacokinetics of bivalirudin have not been studied in patients with hepatic impairment but are not expected to be altered because bivalirudin is not metabolized by liver enzymes such as cytochrome P-450 isozymes.
Renal Insufficiency: The systemic clearance of bivalirudin decreases with glomerular filtration rate (GFR). The clearance of bivalirudin is similar in patients with normal renal function and those with mild renal impairment. Clearance is reduced by approximately 20% in patients with moderate or severe renal impairment, and 80% in dialysis-dependent patients (Table 8).
Table 8 - Pharmacokinetic parameters for bivalirudin in patients with normal and impaired renal function:

In patients with renal insufficiency, coagulation parameters such as the ACT should be monitored during Angiox therapy.
Elderly: Pharmacokinetics have been evaluated in elderly patients as part of a renal pharmacokinetic study. Dose adjustments for this age group should be on the basis of renal function, see section 4.2.
Gender: There are no gender effects in the pharmacokinetics of bivalirudin.
Weight: Bivalirudin dose is body weight adjusted in mg/kg.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety, pharmacology, repeated dose toxicity, genotoxicity, or toxicity to reproduction.
Toxicity in animals upon repeated or continuous exposure (1 day to 4 weeks at exposure levels of up to 10 times the clinical steady state plasma concentration) was limited to exaggerated pharmacological effects. Comparison of the single and repeated dose studies revealed that toxicity was related primarily to duration of exposure. All the undesirable effects, primary and secondary, resulting from excessive pharmacological activity were reversible. Undesirable effects that resulted from prolonged physiological stress in response to a non-homeostatic state of coagulation were not seen after short exposure comparable to that in clinical use, even at much higher doses.
Bivalirudin is intended for short-term administration and therefore no data on the long-term carcinogenic potential of bivalirudin are available. However, bivalirudin was not mutagenic or clastogenic in standard assays for such effects.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.