ALECENSA Hard capsule Ref.[6109] Active ingredients: Alectinib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639 Grenzach-Wyhlen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: anti-neoplastic agents, protein kinase inhibitor
ATC code: L01ED03
Mechanism of action
Alectinib is a highly selective and potent ALK and rearranged during transfection (RET) tyrosine kinase inhibitor. In pre-clinical studies, inhibition of ALK tyrosine kinase activity led to blockage of downstream signalling pathways including signal transducer and activator of transcription 3 (STAT 3) and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and induction of tumour cell death (apoptosis).
Alectinib demonstrated in vitro and in vivo activity against mutant forms of the ALK enzyme, including mutations responsible for resistance to crizotinib. The major metabolite of alectinib (M4) has shown similar in vitro potency and activity.
Based on preclinical data, alectinib is not a substrate of P-gp or BCRP, which are both efflux transporters in the blood brain barrier, and is therefore able to distribute into and be retained within the central nervous system.
Clinical efficacy and safety
Adjuvant treatment of resected ALK-positive NSCLC
The efficacy of Alecensa for the adjuvant treatment of patients with ALK-positive NSCLC following complete tumour resection was established in a global randomised Phase III open-label clinical trial (BO40336; ALINA). Eligible patients were required to have Stage IB (tumours ≥4 cm) - Stage IIIA NSCLC per the Union for International Cancer Control/American Joint Committee on Cancer (UICC/AJCC) Staging System, 7th Edition, with ALK-positive disease identified by a locally performed CE-marked ALK test, or centrally performed by the Ventana ALK (D5F3) immunohistochemistry (IHC) assay.
The following selection criteria define patients with high risk of recurrence who are included in the therapeutic indication and are reflective of the patient population with Stage IB (tumours ≥4 cm) – IIIA NSCLC according to the 7th Edition UICC/AJCC staging criteria:
Tumour size ≥4 cm; or tumours of any size that are either accompanied by N1 or N2 status; or tumours that are invasive of thoracic structures (directly invade the parietal pleura, chest wall, diaphragm, phrenic nerve, mediastinal pleura, parietal pericardium, mediastinum, heart, great vessels, trachea, recurrent laryngeal nerve, oesophagus, vertebral body, carina); or tumours that involve the main bronchus <2 cm distal to the carina but without involvement of the carina; or tumours that are associated with atelectasis or obstructive pneumonitis of the entire lung; or tumours with separate nodule(s) in the same lobe or different ipsilateral lobe as the primary.
The study did not include patients who had N2 status with tumours also invading the mediastinum, heart, great vessels, trachea, recurrent laryngeal nerve, oesophagus, vertebral body, carina, or with separate tumour nodule(s) in a different ipsilateral lobe.
Patients were randomised (1:1) to receive Alecensa or platinum-based chemotherapy following tumour resection. Randomisation was stratified by race (Asian and non-Asian) and stage of disease (IB, II and IIIA). Alecensa was administered at the recommended oral dose of 600 mg twice daily for a total of 2 years, or until disease recurrence or unacceptable toxicity. Platinum-based chemotherapy was administered intravenously for 4 cycles, with each cycle lasting 21 days, according to one of the following regimens:
Cisplatin 75 mg/m² on Day 1 plus vinorelbine 25 mg/m² on Days 1 and 8
Cisplatin 75 mg/m² on Day 1 plus gemcitabine 1250 mg/m² on Days 1 and 8
Cisplatin 75 mg/m² on Day 1 plus pemetrexed 500 mg/m² on Day 1
In the event of intolerance to a cisplatin-based regimen, carboplatin was administered instead of cisplatin in the above combinations at a dose of area under the free carboplatin plasma versus time curve (AUC) 5 mg/mL/min or AUC 6 mg/mL/min.
The primary efficacy endpoint was disease-free survival (DFS) as assessed by the Investigator. DFS was defined as the time from date of randomisation to the date of occurrence of any of the following: first documented recurrence of disease, new primary NSCLC, or death due to any cause, whichever occurred first. The secondary and exploratory efficacy endpoints were overall survival (OS) and time to CNS recurrence or death (CNS-DFS).
A total of 257 patients were studied: 130 patients were randomised to the Alecensa arm, and 127 patients were randomised to the chemotherapy arm. Overall, the median age was 56 years (range: 26 to 87), and 24% were ≥65 years old, 52% were female, 56% were Asian, 60% were never smokers, 53% had an ECOG PS of 0, 10% of patients had Stage IB, 36% had Stage II and 54% had Stage IIIA disease.
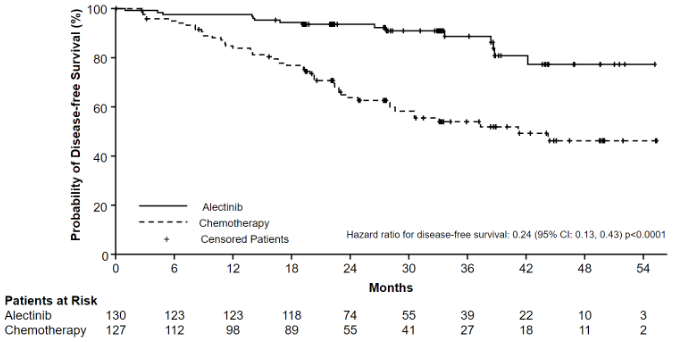
ALINA demonstrated a statistically significant improvement in DFS for patients treated with Alecensa compared to patients treated with chemotherapy in the Stage II-IIIA and the Stage IB (≥4 cm) - IIIA (ITT) patient populations. OS data were not mature at the time of DFS analysis with 2.3% of deaths reported overall. The median duration of survival follow-up was 27.8 months in the Alecensa arm and 28.4 months in the chemotherapy arm.
The DFS efficacy results are summarised in Table 4 and Figure 1.
Table 4. Investigator assessed DFS results in ALINA:
| Efficacy Parameter | Stage II-IIIA | ITT Population | ||
|---|---|---|---|---|
| Alecensa N=116 | Chemotherapy N=115 | Alecensa N=130 | Chemotherapy N=127 | |
| Number of DFS Events (%) | 14 (12.1) | 45 (39.1) | 15 (11.5) | 50 (39.4) |
| Median DFS, months (95% CI) | NE (NE, NE) | 44.4 (27.8, NE) | NE (NE, NE) | 41.3 (28.5, NE) |
| Stratified HR (95% CI)* | 0.24 (0.13, 0.45) | 0.24 (0.13, 0.43) | ||
| p-value (log-rank)* | <0.0001 | <0.0001 | ||
DFS = Disease-Free Survival; ITT = Intent-to-Treat; CI = Confidence Interval; NE = Not Estimable; HR = Hazard Ratio
* Stratified by race in Stage II-IIIA, stratified by race and stage in Stage IB-IIIA.
Figure 1. Kaplan-Meier curve of investigator assessed DFS in the ITT population:
Treatment of advanced ALK-positive NSCLC
Treatment-naïve patients
The safety and efficacy of Alecensa were studied in a global randomised Phase III open label clinical trial (BO28984, ALEX) in ALK-positive NSCLC patients who were treatment naïve. Central testing for ALK protein expression positivity of tissue samples from all patients by Ventana anti-ALK (D5F3) immunohistochemistry was required before randomisation into the study.
A total of 303 patients were included in the Phase III trial, 151 patients randomised to the crizotinib arm and 152 patients randomised to the Alecensa arm receiving Alecensa orally, at the recommended dose of 600 mg twice daily.
Eastern Cooperative Oncology Group performance status ((ECOG PS) (0/1 vs. 2)), race (Asian vs. non-Asian), and central nervous system (CNS) metastases at baseline (yes vs. no) were stratification factors for randomisation. The primary endpoint of the trial was to demonstrate superiority of Alecensa versus crizotinib based on Progression Free survival (PFS) as per investigator assessment using Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Baseline demographic and disease characteristics for Alecensa were median age 58 years (54 years for crizotinib), 55% female (58% for crizotinib), 55% non-Asian (54% for crizotinib), 61% with no smoking history (65% for crizotinib), 93% ECOG PS of 0 or 1 (93% for crizotinib), 97% Stage IV disease (96% for crizotinib), 90% adenocarcinoma histology (94% for crizotinib), 40% CNS metastases at baseline (38% for crizotinib) and 17% having received prior CNS radiation (14% for crizotinib).
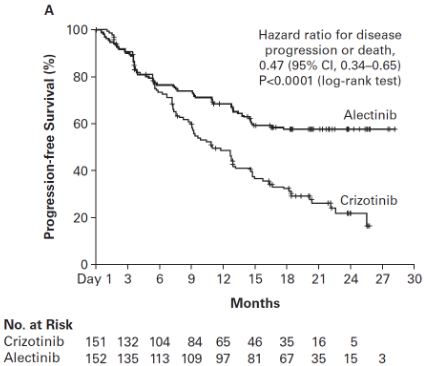
The trial met its primary endpoint at the primary analysis, demonstrating a statistically significant improvement in PFS by investigator. Efficacy data are summarised in Table 5 and the Kaplan-Meier curve for investigator assessed PFS is shown in Figure 2.
Table 5. Summary of efficacy results from study BO28984 (ALEX):
| Crizotinib N=151 | Alecensa N=152 | |
|---|---|---|
| Median duration of follow-up (months) | 17.6 (range 0.3 – 27.0) | 18.6 (range 0.5 – 29.0) |
| Primary efficacy parameter | ||
| PFS (INV) Number of patients with event n (%) Median (months) [95% CI] | 102 (68%) 11.1 [9.1; 13.1] | 62 (41%) NE [17.7; NE] |
| HR [95% CI] Stratified log-rank p-value | 0.47 [0.34, 0.65] p<0.0001 | |
| Secondary efficacy parameters | ||
| PFS (IRC)* Number of patients with event n (%) Median (months) [95% CI] | 92 (61%) 10.4 [7.7; 14.6] | 63 (41%) 25.7 [19.9; NE] |
| HR [95% CI] Stratified log-rank p-value | 0.50 [0.36; 0.70] p<0.0001 | |
| Time to CNS progression (IRC)*,** Number of patients with event n (%) | 68 (45%) | 18 (12%) |
| Cause-specific HR [95% CI] Stratified log-rank p-value | 0.16 [0.10; 0.28] p<0.0001 | |
| 12-month cumulative incidence of CNS progression (IRC) [95% CI] | 41.4% [33.2; 49.4] | 9.4% [5.4; 14.7] |
| ORR (INV)*,*** Responders n (%) [95% CI] | 114 (75.5%) [67.8; 82.1] | 126 (82.9%) [76.0; 88.5] |
| Overall survival* Number of patients with event n (%) Median (months) [95% CI] | 40 (27%) NE [NE; NE] | 35 (23%) NE [NE; NE] |
| HR [95% CI] | 0.76 [0.48; 1.20] | |
| Duration of response (INV) Median (months) [95% CI] | N=114 11.1 [7.9; 13.0] | N=126 NE [NE; NE] |
| CNS-ORR in patients with measurable CNS metastases at baseline CNS responders n (%) [95% CI] CNS-CR n (%) CNS-DOR, median (months) [95% CI] | N=22 11 (50.0%) [28.2; 71.8] 1 (5%) 5.5 [2.1, 17.3] | N=21 17 (81.0%) [58.1; 94.6] 8 (38%) 17.3 [14.8, NE] |
| CNS-ORR in patients with measurable and non-measurable CNS metastases at baseline (IRC) CNS responders n (%) [95% CI] CNS-CR n (%) CNS-DOR, median (months) [95% CI] | N=58 15 (25.9%) [15.3; 39.0] 5 (9%) 3.7 [3.2, 6.8] | N=64 38 (59.4%) [46.4; 71.5] 29 (45%) NE [17.3, NE] |
* Key secondary endpoints part of the hierarchical testing
** Competing risk analysis of CNS progression, systemic progression and death as competing events
*** 2 patients in the crizotinib arm and 6 patients in the alectinib arm had CR
CI = confidence interval; CNS = central nervous system; CR = complete response; DOR = duration of response; HR = hazard ratio; IRC = Independent Review Committee; INV = investigator; NE = not estimable; ORR = objective response rate; PFS = progression free survival
The PFS benefit was consistent for patients with CNS metastases at baseline (hazard ratio (HR) = 0.40, 95% confidence interval (CI): 0.25-0.64, median PFS for Alecensa = not estimable (NE), 95% CI: 9.2-NE, median PFS for crizotinib = 7.4 months, 95%CI: 6.6-9.6) and without CNS metastases at baseline (HR = 0.51, 95% CI: 0.33-0.80, median PFS for Alecensa = NE, 95% CI: NE, NE, median PFS for crizotinib = 14.8 months, 95% CI:10.8-20.3), indicating benefit of Alecensa over crizotinib in both subgroups.
Figure 2. Kaplan Meier plot of INV assessed PFS in BO28984 (ALEX):
rizotinib pre-treated patients
The safety and efficacy of Alecensa in ALK-positive NSCLC patients pre-treated with crizotinib were studied in two Phase I/II clinical trials (NP28673 and NP28761).
NP28673:
Study NP28673 was a Phase I/II single arm, multicentre study conducted in patients with ALK-positive advanced NSCLC who have previously progressed on crizotinib treatment. In addition to crizotinib, patients may have received previous treatment with chemotherapy. A total of 138 patients were included in the phase II part of the study and received Alecensa orally, at the recommended dose of 600 mg twice daily.
The primary endpoint was to evaluate the efficacy of Alecensa by Objective Response Rate (ORR) as per central Independent Review Committee (IRC) assessment using RECIST version 1.1 in the overall population (with and without prior exposure of cytotoxic chemotherapy treatments). The co-primary endpoint was to evaluate the ORR as per central IRC assessment using RECIST 1.1 in patients with prior exposure of cytotoxic chemotherapy treatments. A lower confidence limit for the estimated ORR above the pre-specified threshold of 35% would achieve a statistically significant result.
Patient demographics were consistent with that of a NSCLC ALK positive population. The demographic characteristics of the overall study population were 67% Caucasian, 26% Asian, 56% females, and the median age was 52 years. The majority of patients had no history of smoking (70%). The ECOG PS at baseline was 0 or 1 in 90.6% of patients and 2 in 9.4% of patients. At the time of entry in the study, 99% of patients had stage IV disease, 61% had brain metastases and in 96% of patients tumours were classified as adenocarcinoma. Among patients included in the study, 20% of the patients had previously progressed on crizotinib treatment only, and 80% had previously progressed on crizotinib and at least one chemotherapy treatment.
Study NP28761:
Study NP28761 was a Phase I/II single arm multicentre study conducted in patients with ALK positive advanced NSCLC who have previously progressed on crizotinib treatment. In addition to crizotinib, patients may have received previous treatment with chemotherapy. A total of 87 patients were included in the phase II part of the study and received Alecensa orally, at the recommended dose of 600 mg twice daily.
The primary endpoint was to evaluate the efficacy of Alecensa by ORR as per central IRC assessment using RECIST version 1.1. A lower confidence limit for the estimated ORR above the pre-specified threshold of 35% would achieve a statistically significant result.
Patient demographics were consistent with that of a NSCLC ALK positive population. The demographic characteristics of the overall study population were 84% Caucasian, 8% Asian, 55% females. The median age was 54 years. The majority of patients had no history of smoking (62%). The ECOG PS at baseline was 0 or 1 in 89.7% of patients and 2 in 10.3% of patients. At the time of entry in the study, 99% of patients had stage IV disease, 60% had brain metastases and in 94% of patients tumours were classified as adenocarcinoma. Among the patients included in the study, 26% of the patients had previously progressed on crizotinib treatment only, and 74% had previously progressed on crizotinib and at least one chemotherapy treatment.
The main efficacy results from studies NP28673 and NP28761 are summarised in Table 6. A summary of pooled analysis of CNS endpoints is presented in Table 7.
Table 6. Efficacy results from studies NP28673 and NP28761:
| NP28673 Alecensa 600 mg twice daily | NP28761 Alecensa 600 mg twice daily | |
|---|---|---|
| Median duration of follow-up (months) | 21 (range 1 – 30) | 17 (range 1 – 29) |
| Primary efficacy parameters | ||
| ORR (IRC) in RE population Responders N (%) [95% CI] | N=122a 62 (50.8%) [41.6%, 60.0%] | N=67b 35 (52.2%) [39.7%, 64.6%] |
| ORR (IRC) in patients pre-treated with chemotherapy Responders N (%) [95% CI] | N=96 43 (44.8%) [34.6%, 55.3%] | |
| Secondary efficacy parameters | ||
| DOR (IRC) Number of patients with events N (%) Median (months) [95% CI] | N=62 36 (58.1%) 15.2 [11.2, 24.9] | N=35 20 (57.1%) 14.9 [6.9, NE] |
| PFS (IRC) Number of patients with events N (%) Median duration (months) [95% CI] | N=138 98 (71.0%) 8.9 [5.6, 12.8] | N=87 58 (66.7%) 8.2 [6.3, 12.6] |
CI = confidence interval; DOR = duration of response; IRC = independent review committee; NE = not estimable; ORR = objective response rate; PFS = progression free survival; RE = response evaluable
a 16 patients did not have measurable disease at baseline according to the IRC and were not included in the IRC response evaluable population.
b 20 patients did not have measurable disease at baseline according to the IRC and were not included in the IRC response evaluable population.
ORR results for studies NP28673 and NP28761 were consistent across subgroups of baseline patient characteristics such as age, gender, race, ECOG PS, CNS metastasis and prior chemotherapy use, especially when considering the small number of patients in some subgroups.
Table 7. Summary of the pooled analysis of CNS endpoints from studies NP28673 and NP28761:
| CNS Parameters (NP28673 and NP28761) | Alecensa 600 mg twice daily |
|---|---|
| Patients with measurable CNS lesions at baseline | |
| CNS ORR (IRC) Responders (%) [95% CI] Complete response Partial response | N=50 32 (64.0%) [49.2%, 77.1%] 11 (22.0%) 21 (42.0%) |
| CNS DOR (IRC) Number of patients with events (%) Median (months) [95%CI] | N=32 18 (56.3%) 11.1 [7.6, NE] |
CI = confidence interval; DOR = duration of response; IRC = independent review committee; ORR = objective response rate; NE = not estimable
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Alecensa in all subsets of the paediatric population in lung carcinoma (small cell and non-small cell carcinoma) (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
The pharmacokinetic parameters for alectinib and its major active metabolite (M4) have been characterised in ALK-positive NSCLC patients and healthy subjects. Based on population pharmacokinetic analysis, the geometric mean (coefficient of variation %) steady-state Cmax, Cmin and AUC0-12hr for alectinib were approximately 665 ng/mL (44.3%), 572 ng/mL (47.8%) and 7430 ng*h/mL (45.7%), respectively. The geometric mean steady-state Cmax, Cmin and AUC0-12hr for M4 were approximately 246 ng/mL (45.4%), 222 ng/mL (46.6%) and 2810 ng*h/mL (45.9%), respectively.
Absorption
Following oral administration of 600 mg twice daily under fed conditions in ALK-positive NSCLC patients, alectinib was absorbed reaching Tmax after approximately 4 to 6 hours.
Alectinib steady-state is reached within 7 days with continuous 600 mg twice daily dosing. The accumulation ratio for the twice-daily 600 mg regimen was approximately 6-fold. Population PK analysis supports dose proportionality for alectinib across the dose range of 300 to 900 mg under fed conditions.
The absolute bioavailability of alectinib capsules was 36.9% (90% CI: 33.9%, 40.3%) under fed conditions in healthy subjects.
Following a single oral administration of 600 mg with a high-fat, high-calorie meal, alectinib and M4 exposure was increased by around 3-fold relative to fasted conditions (see section 4.2).
Distribution
Alectinib and its major metabolite M4 are highly bound to human plasma proteins (>99%), independent of active substance concentration. The mean in vitro human blood-to-plasma concentration ratios of alectinib and M4 are 2.64 and 2.50, respectively, at clinically relevant concentrations.
The geometric mean volume of distribution at steady state (Vss) of alectinib following intravenous (IV) administration was 475 L, indicating extensive distribution into tissues. Based on in vitro data, alectinib is not a substrate of P-gp. Alectinib and M4 are not substrates of BCRP or organic anion-transporting polypeptide (OATP) 1B1/B3.
Biotransformation
In vitro metabolism studies showed that CYP3A4 is the main CYP isozyme mediating alectinib and its major metabolite M4 metabolism, and is estimated to contribute 40-50% of alectinib metabolism. Results from the human mass balance study demonstrated that alectinib and M4 were the main circulating moieties in plasma with 76% of the total radioactivity in plasma. The geometric mean Metabolite/Parent ratio at steady state is 0.399.
Metabolite M1b was detected as a minor metabolite from in vitro and in human plasma in healthy subjects. Formation of metabolite M1b and its minor isomer M1a is likely to be catalyzed by a combination of CYP isozymes (including isozymes other than CYP3A) and aldehyde dehydrogenase (ALDH) enzymes.
In vitro studies indicate that neither alectinib nor its major active metabolite (M4) inhibits CYP1A2, CYP2B6, CYP2C9, CYP2C19, or CYP2D6 at clinically relevant concentrations. Alectinib did not inhibit OATP1B1/OATP1B3, OAT1, OAT3 or OCT2 at clinically relevant concentrations in vitro.
Elimination
Following administration of a single dose of 14C-labeled alectinib administered orally to healthy subjects the majority of radioactivity was excreted in faeces (mean recovery 97.8%) with minimal excretion in urine (mean recovery 0.46%). In faeces, 84% and 5.8% of the dose was excreted as unchanged alectinib or M4, respectively.
Based on a population PK analysis, the apparent clearance (CL/F) of alectinib was 81.9 L/hour. The geometric mean of the individual elimination half-life estimates for alectinib was 32.5 hours. The corresponding values for M4 were 217 L/hour and 30.7 hours, respectively.
Pharmacokinetics in special populations
Renal impairment
Negligible amounts of alectinib and the active metabolite M4 are excreted unchanged in urine (<0.2% of the dose). Based on a population pharmacokinetic analysis alectinib and M4 exposures were similar in patients with mild and moderate renal impairment and normal renal function. The pharmacokinetics of alectinib has not been studied in patients with severe renal impairment.
Hepatic impairment
As elimination of alectinib is predominantly through metabolism in the liver, hepatic impairment may increase the plasma concentration of alectinib and/or its major metabolite M4. Based on a population pharmacokinetic analysis, alectinib and M4 exposures were similar in patients with mild hepatic impairment and normal hepatic function.
Following administration of a single oral dose of 300 mg alectinib in subjects with severe (Child-Pugh C) hepatic impairment, alectinib Cmax was the same and AUCinf was 2.2-fold higher compared with the same parameters in matched healthy subjects. M4 Cmax and AUCinf was 39% and 34% lower respectively, resulting in a combined exposure of alectinib and M4 (AUCinf) 1.8-fold higher in patients with severe hepatic impairment compared with matched healthy subjects.
The hepatic impairment study also included a group with moderate (Child-Pugh B) hepatic impairment, and a modestly higher alectinib exposure was observed in this group compared with matched healthy subjects. The subjects in the Child Pugh B group however did in general not suffer from abnormal bilirubin, albumin or prothrombin time, indicating that they may not be fully representative of moderately hepatically impaired subjects with decreased metabolic capacity.
Effects of age, body weight, race and gender
Age, body weight, race and gender had no clinically meaningful effect on the systemic exposure of alectinib and M4. The range of body weights for patients enrolled in clinical studies is 36.9-123 kg. There are no available data on patients with extreme body weight (>130 kg) (see section 4.2).
Preclinical safety data
Carcinogenicity
Carcinogenicity studies have not been performed to establish the carcinogenic potential of alectinib.
Mutagenicity
Alectinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay but induced a slight increase in numerical aberrations in the in vitro cytogenetic assay using Chinese Hamster Lung (CHL) cells with metabolic activation, and micronuclei in a rat bone marrow micronucleus test. The mechanism of micronucleus induction was abnormal chromosome segregation (aneugenicity), and not a clastogenic effect on chromosomes.
Impairment of fertility
No fertility studies in animals have been performed to evaluate the effect of alectinib. No adverse effects on male and female reproductive organs were observed in general toxicology studies. These studies were conducted in rats and monkeys at exposures equal to or greater than 2.6- and 0.5-fold, respectively, of the human exposure, measured by area under the curve (AUC), at the recommended dose of 600 mg twice daily.
Teratogenicity
Alectinib caused embryo-foetal toxicity in pregnant rats and rabbits. In pregnant rats, alectinib caused total embryo-foetal loss (miscarriage) at exposures 4.5-fold of the human AUC exposure and small foetuses with retarded ossification and minor abnormalities of the organs at exposures 2.7-fold of the human AUC exposure. In pregnant rabbits, alectinib caused embryo-foetal loss, small fetuses and increased incidence of skeletal variations at exposures 2.9-fold of the human AUC exposure at the recommended dose.
Other
Alectinib absorbs ultraviolet (UV) light between 200 and 400 nm and demonstrated a phototoxic potential in an in vitro photosafety test in cultured murine fibroblasts after UVA irradiation.
Target organs in both rat and monkey at clinically relevant exposures in the repeat-dose toxicology studies included, but were not limited to the erythroid system, gastrointestinal tract, and hepatobiliary system.
Abnormal erythrocyte morphology was observed at exposures equal or greater than 10-60% the human exposure by AUC at the recommended dose. Proliferative zone extension in gastrointestinal (GI) mucosa in both species was observed at exposures equal to or greater than 20-120% of the human AUC exposure at the recommended dose. Increased hepatic alkaline phosphatase (ALP) and direct bilirubin as well as vacuolation/degeneration/necrosis of bile duct epithelium and enlargement/focal necrosis of hepatocytes was observed in rats and/or monkeys at exposures equal to or greater than 20-30% of the human exposure by AUC at the recommended dose.
A mild hypotensive effect has been observed in monkeys at around clinically relevant exposures.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.