APTIVUS Soft capsules Ref.[8809] Active ingredients: Tipranavir
Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Boehringer Ingelheim International GmbH, Binger Strasse 173, D-55216 Ingelheim am Rhein, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: antivirals for systemic use, protease inhibitors
ATC code: J05AE09
Mechanism of action
The human immunodeficiency virus (HIV-1) encodes an aspartyl protease that is essential for the cleavage and maturation of viral protein precursors. Tipranavir is a non-peptidic inhibitor of the HIV-1 protease that inhibits viral replication by preventing the maturation of viral particles.
Antiviral activity in vitro
Tipranavir inhibits the replication of laboratory strains of HIV-1 and clinical isolates in acute models of T-cell infection, with 50% and 90% effective concentrations (EC50 and EC90) ranging from 0.03 to 0.07 µM (18-42 ng/ml) and 0.07 to 0.18 µM (42-108 ng/ml), respectively. Tipranavir demonstrates antiviral activity in vitro against a broad panel of HIV-1 group M non-clade B isolates (A, C, D, F, G, H, CRF01 AE, CRF02 AG, CRF12 BF). Group O and HIV-2 isolates have reduced susceptibility in vitro to tipranavir with EC50 values ranging from 0.164-1 µM and 0.233-0.522 µM, respectively. Protein binding studies have shown that the antiviral activity of tipranavir decreases on average 3.75-fold in conditions where human serum is present.
Resistance
The development of resistance to tipranavir in vitro is slow and complex. In one particular in vitro resistance experiment, an HIV-1 isolate that was 87-fold resistant to tipranavir was selected after 9 months, and contained 10 mutations in the protease: L10F, I13V, V32I, L33F, M36I, K45I, I54V/T, A71V, V82L, I84V as well as a mutation in the gag polyprotein CA/P2 cleavage site. Reverse genetic experiments showed that the presence of 6 mutations in the protease (I13V, V32I, L33F, K45I, V82L, I84V) was required to confer >10-fold resistance to tipranavir while the full 10-mutation genotype conferred 69-fold resistance to tipranavir. In vitro, there is an inverse correlation between the degree of resistance to tipranavir and the capacity of viruses to replicate. Recombinant viruses showing ≥3-fold resistance to tipranavir grow at less than 1% of the rate detected for wild type HIV-1 in the same conditions. Tipranavir resistant viruses which emerge in vitro from wild-type HIV-1 show decreased susceptibility to the protease inhibitors amprenavir, atazanavir, indinavir, lopinavir, nelfinavir and ritonavir but remain sensitive to saquinavir.
Through a series of multiple stepwise regression analyses of baseline and on-treatment genotypes from all clinical studies, 16 amino acids have been associated with reduced tipranavir susceptibility and/or reduced 48-week viral load response: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D and 84V. Clinical isolates that exhibited a ≥10-fold decrease in tipranavir susceptibility harboured 8 or more tipranavir-associated mutations. In Phase II and III clinical trials, 276 patients with on-treatment genotypes have demonstrated that the predominant emerging mutations with tipranavir treatment are L33F/I/V, V82T/L and I84V. Combination of all three of these is usually required for reduced susceptibility. Mutations at position 82 occur via two pathways: one from preexisting mutation 82A selecting to 82T, the other from wild type 82V selecting to 82L.
Cross-resistance
Tipranavir maintains significant antiviral activity (<4-fold resistance) against the majority of HIV-1 clinical isolates showing post-treatment decreased susceptibility to the currently approved protease inhibitors: amprenavir, atazanavir, indinavir, lopinavir, ritonavir, nelfinavir and saquinavir. Greater than 10-fold resistance to tipranavir is uncommon (<2.5% of tested isolates) in viruses obtained from highly treatment experienced patients who have received multiple peptidic protease inhibitors.
ECG evaluation
The effect of tipranavir with low dose of ritonavir on the QTcF interval was measured in a study in which 81 healthy subjects received the following treatments twice daily for 2.5 days: tipranavir/ritonavir (500/200 mg), tipranavir/ritonavir at a supra-therapeutic dose (750/200 mg), and placebo/ritonavir (-/200 mg). After baseline and placebo adjustment, the maximum mean QTcF change was 3.2 ms (1-sided 95% Upper CI: 5.6 ms) for the 500/200 mg dose and 8.3 ms (1-sided 95% Upper CI: 10.8 ms) for the supra-therapeutic 750/200 mg dose. Hence tipranavir at therapeutic dose with low dose of ritonavir did not prolong the QTc interval but may do so at supratherapeutic dose.
Clinical pharmacodynamic data
This indication is based on the results of two phase III studies, performed in highly pre-treated adult patients (median number of 12 prior antiretroviral agents) with virus resistant to protease inhibitors and of one phase II study investigating pharmacokinetics, safety and efficacy of Aptivus in mostly treatment-experienced adolescent patients aged 12 to 18 years.
The following clinical data is derived from analyses of 48-week data from ongoing studies (RESIST-1 and RESIST-2) measuring effects on plasma HIV RNA levels and CD4 cell counts. RESIST-1 and RESIST-2 are ongoing, randomised, open-label, multicentre studies in HIV-positive, triple-class experienced patients, evaluating treatment with 500 mg tipranavir co-administered with low dose ritonavir (200 mg; twice daily) plus an optimised background regimen (OBR) individually defined for each patient based on genotypic resistance testing and patient history. The comparator regimen included a ritonavir-boosted PI (also individually defined) plus an OBR. The ritonavir-boosted PI was chosen from among saquinavir, amprenavir, indinavir or lopinavir/ritonavir.
All patients had received at least two PI-based antiretroviral regimens and were failing a PI-based regimen at the time of study entry. At least one primary protease gene mutation from among 30N, 46I, 46L, 48V, 50V, 82A, 82F, 82L, 82T, 84V or 90M had to be present at baseline, with not more than two mutations on codons 33, 82, 84 or 90.
After Week 8, patients in the comparator arm who met the protocol defined criteria of initial lack of virologic response had the option of discontinuing treatment and switching over to tipranavir with ritonavir in a separate roll-over study.
The 1483 patients included in the primary analysis had a median age of 43 years (range 17-80), were 86% male, 75% white, 13% black and 1% Asian. In the tipranavir and comparator arms median baseline CD4 cell counts were 158 and 166 cells/mm³, respectively, (ranges 1-1893 and 1-1184 cells/mm³); median baseline plasma HIV-1 RNA was 4.79 and 4.80 log10 copies/ml, respectively (ranges 2.34-6.52 and 2.01-6.76 log10 copies/ml).
Patients had prior exposure to a median of 6 NRTIs, 1 NNRTI, and 4 PIs. In both studies, a total of 67% patient viruses were resistant and 22% were possibly resistant to the pre-selected comparator PIs. A total of 10% of patients had previously used enfuvirtide. Patients had baseline HIV-1 isolates with a median of 16 HIV-1 protease gene mutations, including a median of 3 primary protease gene mutations D30N, L33F/I, V46I/L, G48V, I50V, V82A/F/T/L, I84V, and L90M. With respect to mutations on codons 33, 82, 84 and 90 approximately 4% had no mutations, 24% had mutations at codons 82 (less than 1% of patients had the mutation V82L) and 90, 18% had mutations at codons 84 and 90 and 53% had at least one key mutation at codon 90. One patient in the tipranavir arm had four mutations. In addition the majority of participants had mutations associated with both NRTI and NNRTI resistance. Baseline phenotypic susceptibility was evaluated in 454 baseline patient samples. There was an average decrease in susceptibility of 2-fold wild type (WT) for tipranavir, 12-fold WT for amprenavir, 55-fold WT for atazanavir, 41-fold WT for indinavir, 87-fold WT for lopinavir, 41- fold WT for nelfinavir, 195-fold WT for ritonavir, and 20-fold WT for saquinavir.
Combined 48-week treatment response (composite endpoint defined as patients with a confirmed ≥1 log RNA drop from baseline and without evidence of treatment failure) for both studies was 34% in the tipranavir with ritonavir arm and 15% in the comparator arm. Treatment response is presented for the overall population (displayed by enfuvirtide use), and detailed by PI strata for the subgroup of patients with genotypically resistant strains in the Table below.
Treatment response* at week 48 (pooled studies RESIST-1 and RESIST-2 in treatmentexperienced patients)
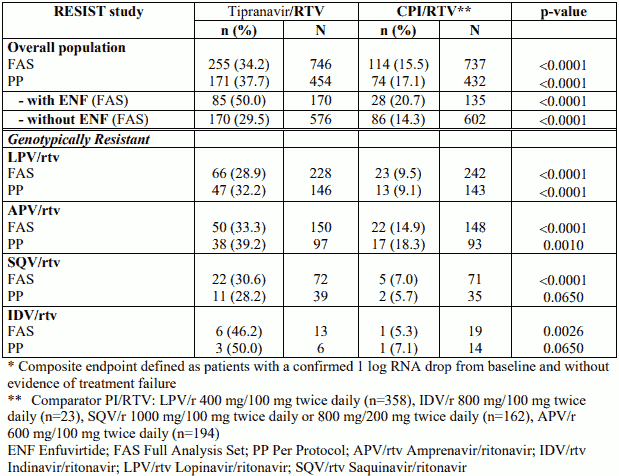
Combined 48-week median time to treatment failure for both studies was 115 days in the tipranavir with ritonavir arm and 0 days in the comparator arm (no treatment response was imputed to day 0).
Through 48 weeks of treatment, the proportion of patients in the tipranavir with ritonavir arm compared to the comparator PI/ritonavir arm with HIV-1 RNA <400 copies/ml was 30% and 14% respectively, and with HIV-1 RNA <50 copies/ml was 23% and 10% respectively. Among all randomised and treated patients, the median change from baseline in HIV-1 RNA at the last measurement up to Week 48 was -0.64 log10 copies/ml in patients receiving tipranavir with ritonavir versus -0.22 log10 copies/ml in the comparator PI/ritonavir arm.
Among all randomised and treated patients, the median change from baseline in CD4+ cell count at the last measurement up to Week 48 was +23 cells/mm³ in patients receiving tipranavir with ritonavir (N=740) versus +4 cells/mm³ in the comparator PI/ritonavir (N=727) arm.
The superiority of tipranavir co-administered with low dose ritonavir over the comparator protease inhibitor/ritonavir arm was observed for all efficacy parameters at week 48. It has not been shown that tipranavir is superior to these boosted comparator protease inhibitors in patients harbouring strains susceptible to these protease inhibitors. RESIST data also demonstrate that tipranavir co-administered with low dose ritonavir exhibits a better treatment response at 48 weeks when the OBR contains genotypically available antiretroviral agents (e.g. enfuvirtide).
At present there are no results from controlled trials evaluating the effect of tipranavir on clinical progression of HIV.
Paediatric population
HIV-positive, paediatric patients, aged 2 through 18 years, were studied in a randomized, open-label, multicenter study (trial 1182.14). Patients were required to have a baseline HIV-1 RNA concentration of at least 1500 copies/ml, were stratified by age (2 to <6 years, 6 to <12 years and 12 to 18 years) and randomized to receive one of two tipranavir with ritonavir dose regimens: 375 mg/m² / 150 mg/m² dose, compared to the 290 mg/m² / 115 mg/m² dose, plus background therapy of at least two nonprotease inhibitor antiretroviral medicinal products, optimized using baseline genotypic resistance testing. All patients initially received Aptivus oral solution. Paediatric patients who were 12 years or older and received the maximum dose of 500 mg/200 mg twice daily could change to Aptivus capsules from study day 28. The trial evaluated pharmacokinetics, safety and tolerability, as well as virologic and immunologic responses through 48 weeks.
No data are available on the efficacy and safety of Aptivus capsules in children less than 12 years of age. Since Aptivus capsules and oral solution are not bioequivalent, results obtained with the oral solution cannot be extrapolated to the capsules (see also section 5.2). In patients with a body surface area of less than 1.33 m² appropriate dose adjustments cannot be achieved with the capsule formulation.
The baseline characteristics and the key efficacy results at 48 weeks for the paediatric patients receiving Aptivus capsules are displayed in the tables below. Data on the 29 patients who switched to capsules during the first 48 weeks are presented. Due to limitations in the study design (e.g. nonrandomized switch allowed according to patient/clinician decision), any comparisons between patients taking capsules and oral solution are not meaningful.
Baseline characteristics for patients 12-18 years of age who took capsule
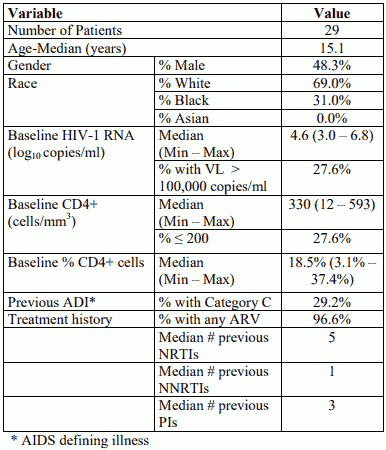
Key efficacy results at 48 weeks for patients 12-18 years of age who took capsule
| Endpoint | Result |
|---|---|
| Number of patients | 29 |
| Primary efficacy endpoint: % with VL <400 | 31.0% |
| Median change from baseline in log10 HIV-1 RNA (copies/ml) | -0.79 |
| Median change from baseline in CD4+ cell count (cells/mm3) | 39 |
| Median change from baseline in % CD4+ cells | 3% |
Analyses of tipranavir resistance in treatment experienced patients
Tipranavir with ritonavir response rates in the RESIST studies were assessed by baseline tipranavir genotype and phenotype. Relationships between baseline phenotypic susceptibility to tipranavir, primary PI mutations, protease mutations at codons 33, 82, 84 and 90, tipranavir resistance-associated mutations, and response to tipranavir with ritonavir therapy were assessed.
Of note, patients in the RESIST studies had a specific mutational pattern at baseline of at least one primary protease gene mutation among codons 30N, 46I, 46L, 48V, 50V, 82A, 82F, 82L, 82T, 84V or 90M, and no more than two mutations on codons 33, 82, 84 or 90.
The following observations were made:
Primary PI mutations
Analyses were conducted to assess virological outcome by the number of primary PI mutations (any change at protease codons 30, 32, 36, 46, 47, 48, 50, 53, 54, 82, 84, 88 and 90) present at baseline. Response rates were higher in tipranavir with ritonavir patients than comparator PI boosted with ritonavir in new enfuvirtide patients, or patients without new enfuvirtide. However, without new enfuvirtide some patients began to lose antiviral activity between weeks 4 and 8.
Mutations at protease codons 33, 82, 84 and 90
A reduced virological response was observed in patients with viral strains harbouring two or more mutations at HIV protease codons 33, 82, 84 or 90, and not receiving new enfuvirtide.
Tipranavir resistance-associated mutations
Virological response to tipranavir with ritonavir therapy has been evaluated using a tipranavirassociated mutation score based on baseline genotype in RESIST-1 and RESIST-2 patients. This score (counting the 16 amino acids that have been associated with reduced tipranavir susceptibility and/or reduced viral load response: 10V, 13V, 20M/R/V, 33F, 35G, 36I, 43T, 46L, 47V, 54A/M/V, 58E, 69K, 74P, 82L/T, 83D and 84V) was applied to baseline viral protease sequences. A correlation between the tipranavir mutation score and response to tipranavir with ritonavir therapy at week 48 has been established.
This score has been determined from the selected RESIST patient population having specific mutation inclusion criteria and therefore extrapolation to a wider population mandates caution.
At 48-weeks, a higher proportion of patients receiving tipranavir with ritonavir achieved a treatment response in comparison to the comparator protease inhibitor/ritonavir for nearly all of the possible combinations of genotypic resistance mutations (see table below).
Proportion of patients achieving treatment response at Week 48 (confirmed ≥1 log10 copies/ml decrease in viral load compared to baseline), according to tipranavir baseline mutation score and enfuvirtide use in RESIST patients
| New ENF | No New ENF* | |
|---|---|---|
| Number of TPV Score Mutations** | TPV/r | TPV/r |
| 0,1 | 73% | 53% |
| 2 | 61% | 33% |
| 3 | 75% | 27% |
| 4 | 59% | 23% |
| ≥5 | 47% | 13% |
| All patients | 61% | 29% |
* Includes patients who did not receive ENF and those who were previously treated with and continued ENF
** Mutations in HIV protease at positions L10V, I13V, K20M/R/V, L33F, E35G, M36I, K43T, M46L, I47V, I54A/M/V, 58E, H69K, T74P, V82L/T, N83D or I84V ENF Enfuvirtide; TPV/r Tipranavir with ritonavir
Sustained HIV-1 RNA decreases up to week 48 were mainly observed in patients who received tipranavir with ritonavir and new enfuvirtide. If patients did not receive tipranavir with ritonavir with new enfuvirtide, diminished treatment responses at week 48 were observed, relative to new enfuvirtide use (see Table below).
Mean decrease in viral load from baseline to week 48, according to tipranavir baseline mutation score and enfuvirtide use in RESIST patients
| New ENF | No New ENF* | |
|---|---|---|
| Number of TPV Score Mutations** | TPV/r | TPV/r |
| 0,1 | -2.3 | -1.6 |
| 2 | -2.1 | -1.1 |
| 3 | -2.4 | -0.9 |
| 4 | -1.7 | -0.8 |
| ≥5 | -1.9 | -0.6 |
| All patients | -2.0 | -1.0 |
* Includes patients who did not receive ENF and those who were previously treated with and continued ENF
** Mutations in HIV protease at positions L10V, I13V, K20M/R/V, L33F, E35G, M36I, K43T, M46L, I47V, I54A/M/V, 58E, H69K, T74P, V82L/T, N83D or I84V ENF Enfuvirtide; TPV/r Tipranavir with ritonavir
Tipranavir phenotypic resistance
Increasing baseline phenotypic fold change to tipranavir in isolates is correlated to decreasing virological response. Isolates with baseline fold change of >0 to 3 are considered susceptible; isolates with >3 to 10 fold changes have decreased susceptibility; isolates with >10 fold changes are resistant.
Conclusions regarding the relevance of particular mutations or mutational patterns are subject to change with additional data, and it is recommended to always consult current interpretation systems for analysing resistance test results.
Pharmacokinetic properties
In order to achieve effective tipranavir plasma concentrations and a twice daily dosing regimen, coadministration of tipranavir with low dose ritonavir twice daily is essential (see section 4.2). Ritonavir acts by inhibiting hepatic cytochrome P450 CYP3A, the intestinal P-glycoprotein (P-gp) efflux pump and possibly intestinal cytochrome P450 CYP3A as well. As demonstrated in a doseranging evaluation in 113 HIV-negative healthy male and female volunteers, ritonavir increases AUC0-12h, Cmax and Cmin and decreases the clearance of tipranavir. 500 mg Tipranavir co-administered with low dose ritonavir (200 mg; twice daily) was associated with a 29-fold increase in the geometric mean morning steady-state trough plasma concentrations compared to tipranavir 500 mg twice daily without ritonavir.
Absorption
Absorption of tipranavir in humans is limited, though no absolute quantification of absorption is available. Tipranavir is a P-gp substrate, a weak P-gp inhibitor and appears to be a potent P-gp inducer as well. Data suggest that, although ritonavir is a P-gp inhibitor, the net effect of Aptivus, coadministered with low dose ritonavir, at the proposed dose regimen at steady-state, is P-gp induction. Peak plasma concentrations are reached within 1 to 5 hours after dose administration depending upon the dosage used. With repeated dosing, tipranavir plasma concentrations are lower than predicted from single dose data, presumably due to hepatic enzyme induction. Steady-state is attained in most subjects after 7 days of dosing. Tipranavir, co-administered with low dose ritonavir, exhibits linear pharmacokinetics at steady state.
Dosing with Aptivus capsules 500 mg twice daily concomitant with 200 mg ritonavir twice daily for 2 to 4 weeks and without meal restriction produced a mean tipranavir peak plasma concentration (Cmax) of 94.8 ± 22.8 µM for female patients (n=14) and 77.6 ± 16.6 µM for male patients (n=106), occurring approximately 3 hours after administration. The mean steady-state trough concentration prior to the morning dose was 41.6 ± 24.3 µM for female patients and 35.6 ± 16.7 µM for male patients. Tipranavir AUC over a 12 hour dosing interval averaged 851 ± 309 µM•h (CL=1.15 l/h) for female patients and 710 ± 207 µM•h (CL=1.27 l/h) for male patients. The mean half-life was 5.5 (females) or 6.0 hours (males).
Effects of food on oral absorption
Food improves the tolerability of tipranavir with ritonavir. Therefore Aptivus, co-administered with low dose ritonavir, should be given with food.
Absorption of tipranavir, co-administered with low dose ritonavir, is reduced in the presence of antacids (see section 4.5).
Distribution
Tipranavir is extensively bound to plasma proteins (>99.9%). From clinical samples of healthy volunteers and HIV-1 positive subjects who received tipranavir without ritonavir the mean fraction of tipranavir unbound in plasma was similar in both populations (healthy volunteers 0.015% 0.006%; HIV-positive subjects 0.019% 0.076%). Total plasma tipranavir concentrations for these samples ranged from 9 to 82 M. The unbound fraction of tipranavir appeared to be independent of total concentration over this concentration range.
No studies have been conducted to determine the distribution of tipranavir into human cerebrospinal fluid or semen.
Biotransformation
In vitro metabolism studies with human liver microsomes indicated that CYP3A4 is the predominant CYP isoform involved in tipranavir metabolism.
The oral clearance of tipranavir decreased after the addition of ritonavir which may represent diminished first-pass clearance of the substance at the gastrointestinal tract as well as the liver.
The metabolism of tipranavir in the presence of low dose ritonavir is minimal. In a 14C-tipranavir human study (500 mg 14C-tipranavir with 200 mg ritonavir, twice daily), unchanged tipranavir was predominant and accounted for 98.4% or greater of the total plasma radioactivity circulating at 3, 8, or 12 hours after dosing. Only a few metabolites were found in plasma, and all were at trace levels (0.2% or less of the plasma radioactivity). In faeces, unchanged tipranavir represented the majority of faecal radioactivity (79.9% of faecal radioactivity). The most abundant faecal metabolite, at 4.9% of faecal radioactivity (3.2% of dose), was a hydroxyl metabolite of tipranavir. In urine, unchanged tipranavir was found in trace amounts (0.5% of urine radioactivity). The most abundant urinary metabolite, at 11.0% of urine radioactivity (0.5% of dose) was a glucuronide conjugate of tipranavir.
Elimination
Administration of 14C-tipranavir to subjects (n=8) that received 500 mg tipranavir with 200 mg ritonavir; twice daily dosed to steady-state demonstrated that most radioactivity (median 82.3%) was excreted in faeces, while only a median of 4.4% of the radioactive dose administered was recovered in urine. In addition, most radioactivity (56%) was excreted between 24 and 96 hours after dosing. The effective mean elimination half-life of tipranavir with ritonavir in healthy volunteers (n=67) and HIV-infected adult patients (n=120) was approximately 4.8 and 6.0 hours, respectively, at steady state following a dose of 500 mg/200 mg twice daily with a light meal.
Special populations
Although data available at this stage are currently limited to allow a definitive analysis, they suggest that the pharmacokinetic profile is unchanged in older people and comparable between races. By contrast, evaluation of the steady-state plasma tipranavir trough concentrations at 10-14 h after dosing from the RESIST-1 and RESIST-2 studies demonstrate that females generally had higher tipranavir concentrations than males. After four weeks of Aptivus 500 mg with 200 mg ritonavir (twice daily) the median plasma trough concentration of tipranavir was 43.9 µM for females and 31.1 µM for males. This difference in concentrations does not warrant a dose adjustment.
Renal impairment
Tipranavir pharmacokinetics have not been studied in patients with renal impairment. However, since the renal clearance of tipranavir is negligible, a decrease in total body clearance is not expected in patients with renal impairment.
Hepatic impairment
In a study comparing 9 patients with mild (Child-Pugh A) hepatic impairment to 9 controls, the single and multiple dose exposure of tipranavir and ritonavir were increased in patients with hepatic impairment but still within the range observed in clinical studies. No dosing adjustment is required in patients with mild hepatic impairment but patients should be closely monitored (see sections 4.2 and 4.4).
The influence of moderate (Child-Pugh B) or severe (Child-Pugh C) hepatic impairment on the multiple dose pharmacokinetics of either tipranavir or ritonavir has so far not been investigated. tipranavir is contraindicated in moderate or severe hepatic impairment (see sections 4.2 and 4.3).
Paediatric population
The oral solution has been shown to have greater bioavailability than the soft capsule formulation.
Preclinical safety data
Animal toxicology studies have been conducted with tipranavir alone, in mice, rats and dogs, and coadministered with ritonavir (3.75:1 w/w ratio) in rats and dogs. Studies with co-administration of tipranavir and ritonavir did not reveal any additional toxicological effects when compared to those seen in the tipranavir single agent toxicological studies.
The predominant effects of repeated administration of tipranavir across all species toxicologically tested were on the gastrointestinal tract (emesis, soft stool, diarrhoea) and the liver (hypertrophy). The effects were reversible with termination of treatment. Additional changes included bleeding in rats at high doses (rodents specific). Bleeding observed in rats was associated with prolonged prothrombin time (PT), activated partial thromboplastin time (APTT) and a decrease in some vitamin K dependent factors. The co-administration of tipranavir with vitamin E in the form of TPGS (d-alphatocopherol polyethylene glycol 1000 succinate) from 2,322 IU/m² upwards in rats resulted in a significant increase in effects on coagulation parameters, bleeding events and death. In preclinical studies of tipranavir in dogs, an effect on coagulation parameters was not seen. Co-administration of tipranavir and vitamin E has not been studied in dogs.
The majority of the effects in repeat-dose toxicity studies appeared at systemic exposure levels which are equivalent to or even below the human exposure levels at the recommended clinical dose.
In in vitro studies, tipranavir was found to inhibit platelet aggregation when using human platelets (see section 4.4) and thromboxane A2 binding in an in vitro cell model at levels consistent with exposure observed in patients receiving Aptivus with ritonavir. The clinical implications of these findings are not known.
In a study conducted in rats with tipranavir at systemic exposure levels (AUC) equivalent to human exposure at the recommended clinical dose, no adverse effects on mating or fertility were observed. At maternal doses producing systemic exposure levels similar to or below those at the recommended clinical dose, tipranavir did not produce teratogenic effects. At tipranavir exposures in rats at 0.8-fold human exposure at the clinical dose, foetal toxicity (decreased sternebrae ossification and body weights) was observed. In pre- and post-natal development studies with tipranavir in rats, growth inhibition of pups was observed at maternally toxic doses approximating 0.8-fold human exposure.
Carcinogenicity studies of tipranavir in mice and rats revealed tumourigenic potential specific for these species, which are regarded as of no clinical relevance. Tipranavir showed no evidence of genetic toxicity in a battery of in vitro and in vivo tests.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.