APTIVUS Soft capsules Ref.[8809] Active ingredients: Tipranavir
Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Boehringer Ingelheim International GmbH, Binger Strasse 173, D-55216 Ingelheim am Rhein, Germany
Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Patients with moderate or severe (Child-Pugh B or C) hepatic impairment.
Combination of rifampicin with Aptivus with concomitant low dose ritonavir is contraindicated (see section 4.5).
Herbal preparations containing St John's wort (Hypericum perforatum) due to the risk of decreased plasma concentrations and reduced clinical effects of tipranavir (see section 4.5).
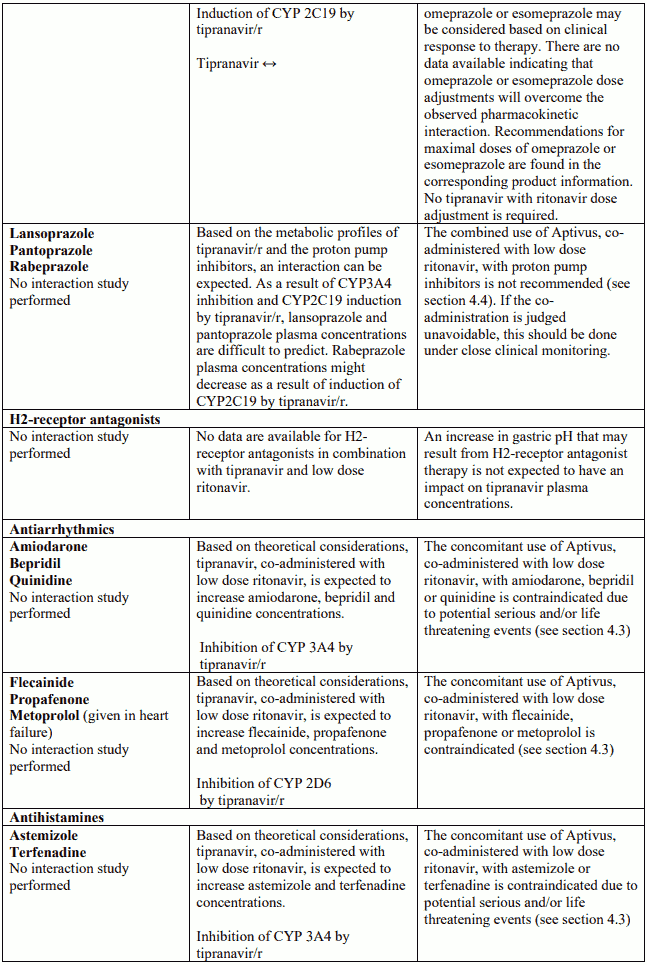
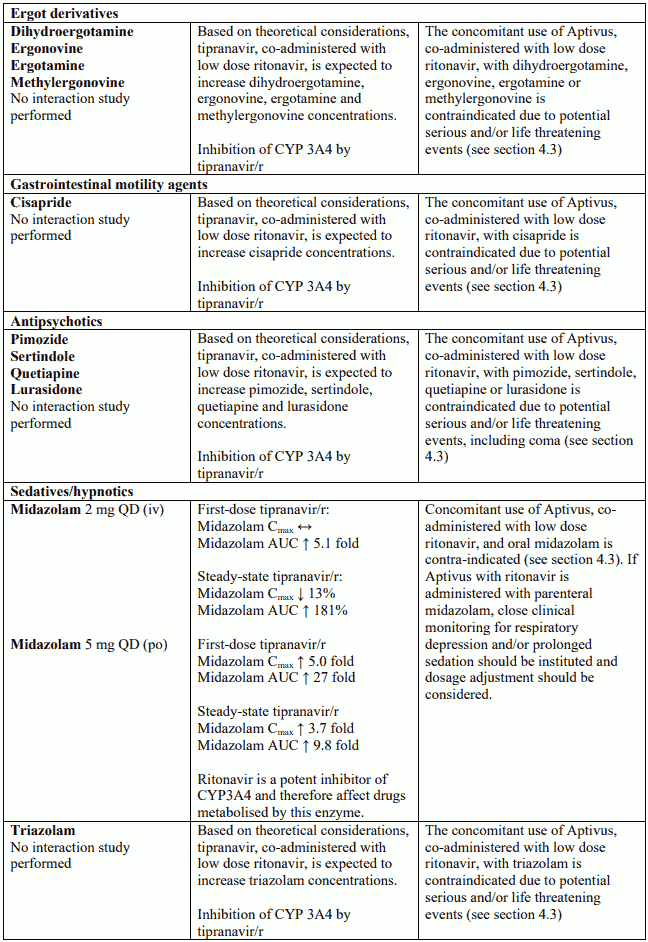
Co-administration of Aptivus with low dose ritonavir, with active substances that are highly dependent on CYP3A for clearance, and for which elevated plasma concentrations are associated with serious and/or life-threatening events. These active substances include antiarrhythmics (such as amiodarone, bepridil, quinidine), antihistamines (such as astemizole, terfenadine), ergot derivatives (such as dihydroergotamine, ergonovine, ergotamine, methylergonovine), gastrointestinal motility agents (such as cisapride), antipsychotics (such as pimozide, sertindole, quetiapine, lurasidone), sedatives/hypnotics (such as orally administered midazolam and triazolam) and HMG-CoA reductase inhibitors (such as simvastatin and lovastatin) (see section 4.5). Also the use of the alpha-1 adrenoceptor antagonist alfuzosin, and sildenafil when used for the treatment of pulmonary arterial hypertension. In addition, co-administration of Aptivus with low dose ritonavir, and medicinal products that are highly dependent on CYP2D6 for clearance, such as the antiarrhythmics flecainide, propafenone and metoprolol given in heart failure (see section 4.5).
Co-administration of colchicine with Aptivus/ritonavir in patients with renal or hepatic impairment (see section 4.5).
Special warnings and precautions for use
Aptivus must be administered with low dose ritonavir to ensure its therapeutic effect (see section 4.2). Failure to correctly co-administer tipranavir with ritonavir will result in reduced plasma levels of tipranavir that may be insufficient to achieve the desired antiviral effect. Patients should be instructed accordingly.
Aptivus is not a cure for HIV-1 infection or AIDS. Patients receiving Aptivus or any other antiretroviral therapy may continue to develop opportunistic infections and other complications of HIV-1 infection.
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Switching from Aptivus capsules to the oral solution
Aptivus capsules are not interchangeable with the oral solution. Compared to the capsules, tipranavir exposure is higher when administering the same dose as oral solution. Also, the composition of the oral solution is different from that of the capsules, with the high vitamin E content being especially noteworthy. Both of these factors may contribute to an increased risk of adverse reactions (type, frequency and/or severity). Therefore patients should not be switched from Aptivus capsules to Aptivus oral solution (see sections 5.1 and 5.2).
Switching from Aptivus oral solution to the capsules
Aptivus oral solution is not interchangeable with the capsules. Compared to the oral solution, tipranavir exposure is lower when administering the same dose as capsules. However, children previously treated with Aptivus oral solution and becoming 12 years of age should be switched to capsules, particularly because of the more favourable safety profile of the capsules. It has to be noted that the switch from the oral solution to the capsule formulation of Aptivus could be associated with decreased exposure. Therefore, it is recommended that patients switching from Aptivus oral solution to capsules at the age of 12 years are closely monitored for the virologic response of their antiretroviral regimen (see sections 5.1 and 5.2).
Liver disease
Aptivus is contraindicated in patients with moderate or severe (Child-Pugh Class B or C) hepatic insufficiency. Limited data are currently available for the use of Aptivus, co-administered with low dose ritonavir, in patients co-infected with hepatitis B or C. Patients with chronic hepatitis B or C and treated with combination antiretroviral therapy are at an increased risk for severe and potentially fatal hepatic adverse reaction. Aptivus should be used in this patient population only if the potential benefit outweighs the potential risk, and with increased clinical and laboratory monitoring. In the case of concomitant antiviral therapy for hepatitis B or C, please refer also to the relevant Summary of Product Characteristics for these medicinal products.
Patients with mild hepatic impairment (Child-Pugh Class A) should be closely monitored.
Patients with pre-existing liver dysfunction including chronic active hepatitis have an increased frequency of liver function abnormalities during combination therapy and should be monitored according to standard practice. Aptivus with ritonavir should be discontinued once signs of worsening liver function occur in patients with pre-existing liver disease.
Aptivus co-administered with low dose ritonavir, has been associated with reports of clinical hepatitis and hepatic decompensation, including some fatalities. These have generally occurred in patients with advanced HIV disease taking multiple concomitant medicinal products. Caution should be exercised when administering Aptivus to patients with liver enzyme abnormalities or with a history of hepatitis. Increased ALAT/ASAT monitoring should be considered in these patients.
Aptivus therapy should not be initiated in patients with pre-treatment ASAT or ALAT greater than 5 times the Upper Limit Normal (ULN) until baseline ASAT/ALAT is stabilised at less than 5X ULN, unless the potential benefit justifies the potential risk.
Aptivus therapy should be discontinued in patients experiencing ASAT or ALAT elevations greater than 10X ULN, or developing signs or symptoms of clinical hepatitis during therapy. If another cause is identified (eg acute hepatitis A, B or C virus, gallbladder disease, other medicinal products), then rechallenge with Aptivus may be considered when ASAT/ALAT have returned to the patient's baseline levels.
Liver monitoring
Monitoring of hepatic tests should be done prior to initiation of therapy, after two, four and then every four weeks until 24 weeks, and then every eight to twelve weeks thereafter. Increased monitoring (i.e. prior to initiation of therapy, every two weeks during the first three months of treatment, then monthly until 48 weeks, and then every eight to twelve weeks thereafter) is warranted when Aptivus and low dose ritonavir are administered to patients with elevated ASAT and ALAT levels, mild hepatic impairment, chronic hepatitis B or C, or other underlying liver disease.
Treatment-naïve patients
In a study performed in antiretroviral naïve adult patients, tipranavir 500 mg with ritonavir 200 mg twice daily, as compared to lopinavir/ritonavir, was associated with an excess in the occurrence of significant (grade 3 and 4) transaminase elevations without any advantage in terms of efficacy (trend towards a lower efficacy). The study was prematurely stopped after 60 weeks. Therefore, tipranavir with ritonavir should not be used in treatment-naïve patients (see section 4.2).
Renal impairment
Since the renal clearance of tipranavir is negligible, increased plasma concentrations are not expected in patients with renal impairment.
Haemophilia
There have been reports of increased bleeding, including spontaneous skin haematomas and haemarthrosis in patients with haemophilia type A and B treated with protease inhibitors. In some patients additional Factor VIII was given. In more than half of the reported cases, treatment with protease inhibitors was continued or reintroduced if treatment had been discontinued. A causal relationship has been evoked, although the mechanism of action had not been elucidated. Haemophiliac patients should therefore be made aware of the possibility of increased bleeding.
Bleeding
RESIST participants receiving Aptivus with ritonavir tended to have an increased risk of bleeding; at 24 weeks the relative risk was 1.98 (95% CI=1.03, 3.80). At 48-weeks the relative risk decreased to 1.27 (95% CI=0.76, 2.12). There was no pattern for the bleeding events and no difference between treatment groups in coagulation parameters. The significance of this finding is being further monitored.
Fatal and non-fatal intracranial haemorrhages (ICH) have been reported in patients receiving Aptivus, many of whom had other medical conditions or were receiving concomitant medicinal products that may have caused or contributed to these events. However, in some cases the role of Aptivus cannot be excluded. No pattern of abnormal haematological or coagulation parameters has been observed in patients in general, or preceding the development of ICH. Therefore, routine measurement of coagulation parameters is not currently indicated in the management of patients on Aptivus.
An increased risk of ICH has previously been observed in patients with advanced HIV disease/AIDS such as those treated in the Aptivus clinical trials.
In in vitro experiments, tipranavir was observed to inhibit human platelet aggregation at levels consistent with exposures observed in patients receiving Aptivus with ritonavir.
In rats, co-administration with vitamin E increased the bleeding effects of tipranavir (see section 5.3).
Aptivus, co-administered with low dose ritonavir, should be used with caution in patients who may be at risk of increased bleeding from trauma, surgery or other medical conditions, or who are receiving medicinal products known to increase the risk of bleeding such as antiplatelet agents and anticoagulants or who are taking supplemental vitamin E. Based on the limits of exposure available from observation in clinical trials, it is recommended not to co-administer to patients more than 1,200 IU vitamin E per day.
Weight and metabolic parameters
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. A higher increase of blood lipids were seen with tipranavir/ritonavir than with comparators (other protease inhibitors) in clinical trials. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Immune reactivation syndrome
In HIV-infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections and pneumocystis pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary. In addition, reactivation of herpes simplex and herpes zoster has been observed in clinical studies with Aptivus, co-administered with low dose ritonavir.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Rash
Mild to moderate rashes including urticarial rash, maculopapular rash, and photosensitivity have been reported in subjects receiving Aptivus, co-administered with low dose ritonavir. At 48-weeks in Phase III trials, rash of various types was observed in 15.5% males and 20.5% females receiving Aptivus coadministered with low dose ritonavir. Additionally, in one interaction trial, in healthy female volunteers administered a single dose of ethinyl oestradiol followed by Aptivus co-administered with low dose ritonavir, 33% of subjects developed a rash. Rash accompanied by joint pain or stiffness, throat tightness, or generalized pruritus has been reported in both men and women receiving Aptivus co-administered with low dose ritonavir. In the paediatric clinical trial, the frequency of rash (all grades, all causality) through 48 weeks of treatment was higher than in adult patients.
Osteonecrosis
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Interactions
The interaction profile of tipranavir, co-administered with low dose ritonavir, is complex. The mechanisms and potential mechanisms contributing to the interaction profile of tipranavir are described (see section 4.5).
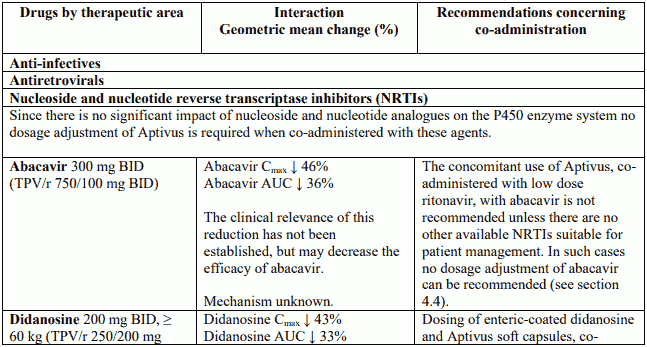
Abacavir and zidovudine
The concomitant use of Aptivus, co-administered with low dose ritonavir, with zidovudine or abacavir, results in a significant decrease in plasma concentration of these nucleoside reverse transcriptase inhibitors (NRTIs). Therefore, the concomitant use of zidovudine or abacavir with Aptivus, co-administered with low dose ritonavir, is not-recommended unless there are no other available NRTIs suitable for patient management (see section 4.5).
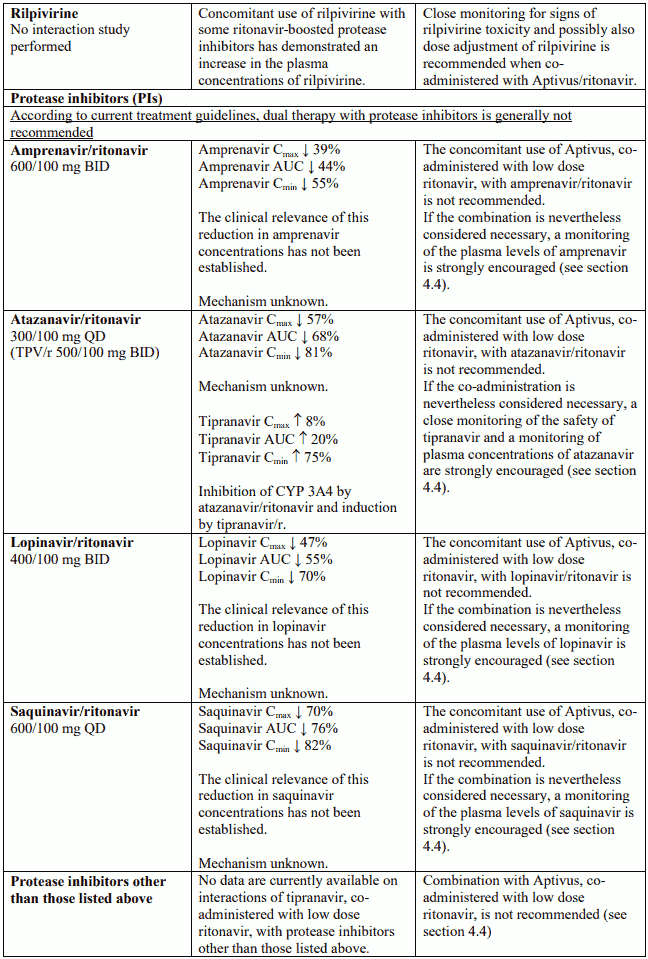
Protease inhibitors
Concomitant use of Aptivus, co-administered with low dose ritonavir, with the protease inhibitors amprenavir, lopinavir or saquinavir (each co-administered with low dose ritonavir) in a dual-boosted regimen, results in significant decreases in plasma concentrations of these protease inhibitors. A significant decrease in plasma concentrations of atazanavir and a marked increase of tipranavir and ritonavir concentrations was observed when Aptivus, associated with low dose ritonavir, was coadministered with atazanavir (see section 4.5). No data are currently available on interactions of tipranavir, co-administered with low dose ritonavir, with protease inhibitors other than those listed above. Therefore, the co-administration of tipranavir, co-administered with low dose ritonavir, with protease inhibitors is not recommended.
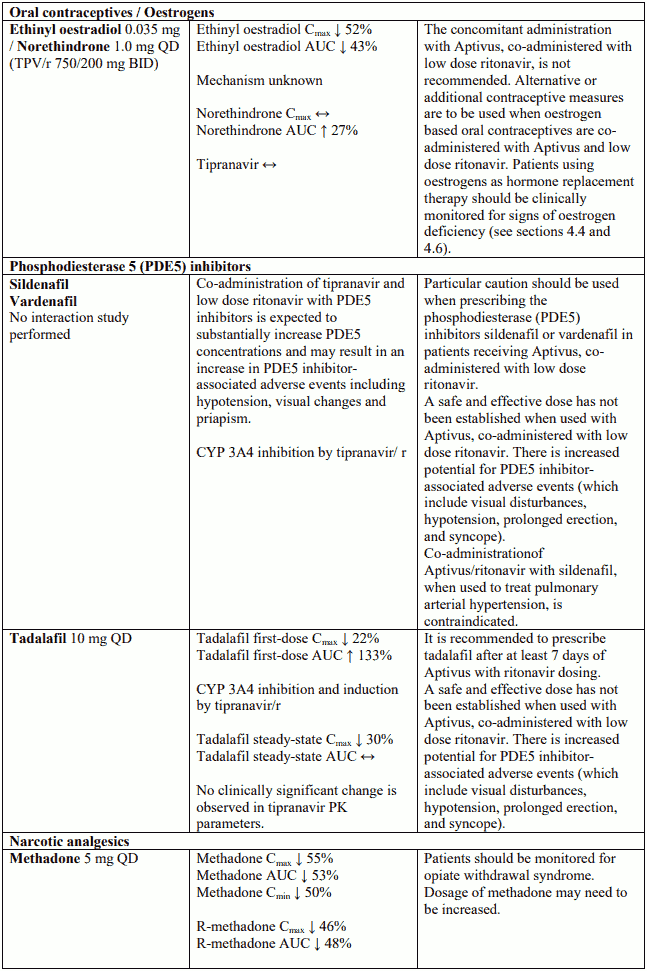
Oral contraceptives and oestrogens
Since levels of ethinyl oestradiol are decreased, the co-administration of Aptivus co-administered with low dose ritonavir is not recommended. Alternative or additional contraceptive measures are to be used when oestrogen based oral contraceptives are co-administered with Aptivus co-administered with low dose ritonavir (see section 4.5). Patients using oestrogens as hormone replacement therapy should be clinically monitored for signs of oestrogen deficiency. Women using oestrogens may have an increased risk of non serious rash.
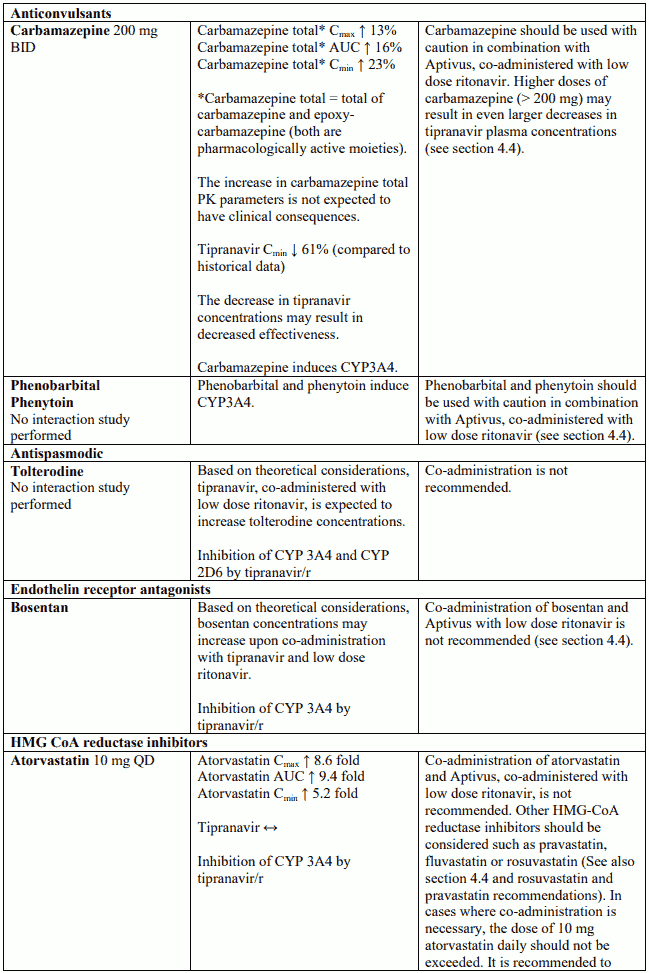
Anticonvulsants
Caution should be used when prescribing carbamazepine, phenobarbital, and phenytoin. Aptivus may be less effective due to decreased tipranavir plasma concentrations in patients taking these agents concomitantly.
Halofantrine, lumefantrine
Due to their metabolic profile and inherent risk of inducing torsades de pointes, administration of halofantrine and lumefantrine with Aptivus co-administered with low dose ritonavir, is not recommended.
Disulfiram / metronidazole
Aptivus soft capsules contain alcohol (7% ethanol, ie 100 mg per capsule or up to 200 mg per dose) which can produce disulfiram-like reactions when co-administered with disulfiram or other medicinal products which produce this reaction (e.g. metronidazole).
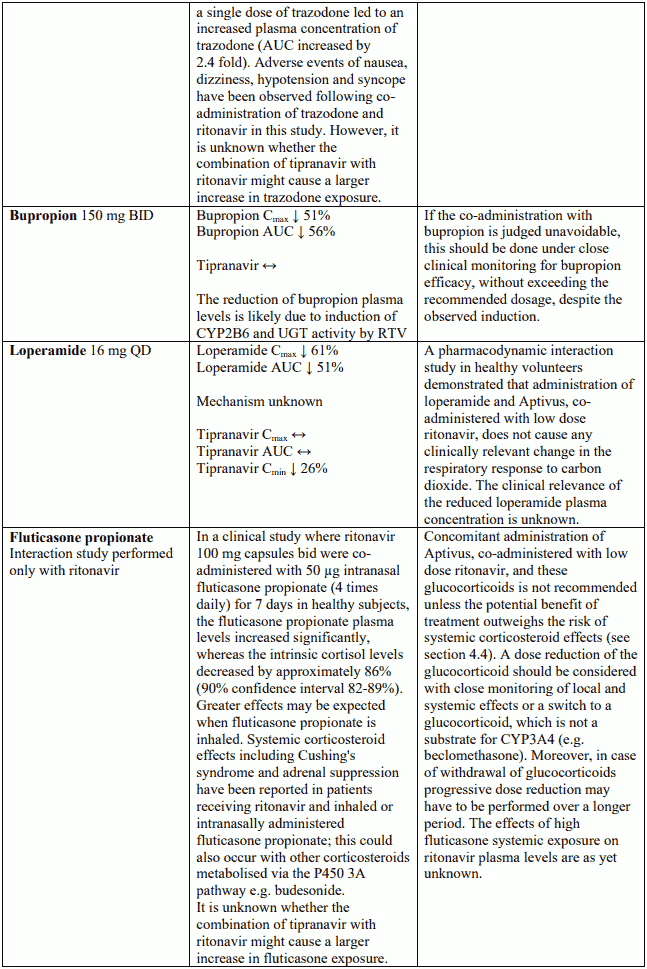
Fluticasone
Concomitant use of tipranavir, co-administered with low dose ritonavir, and fluticasone or other glucocorticoids that are metabolised by CYP3A4 is not recommended unless the potential benefit of treatment outweighs the risk of systemic corticosteroid effects, including Cushing's syndrome and adrenal suppression (see section 4.5).
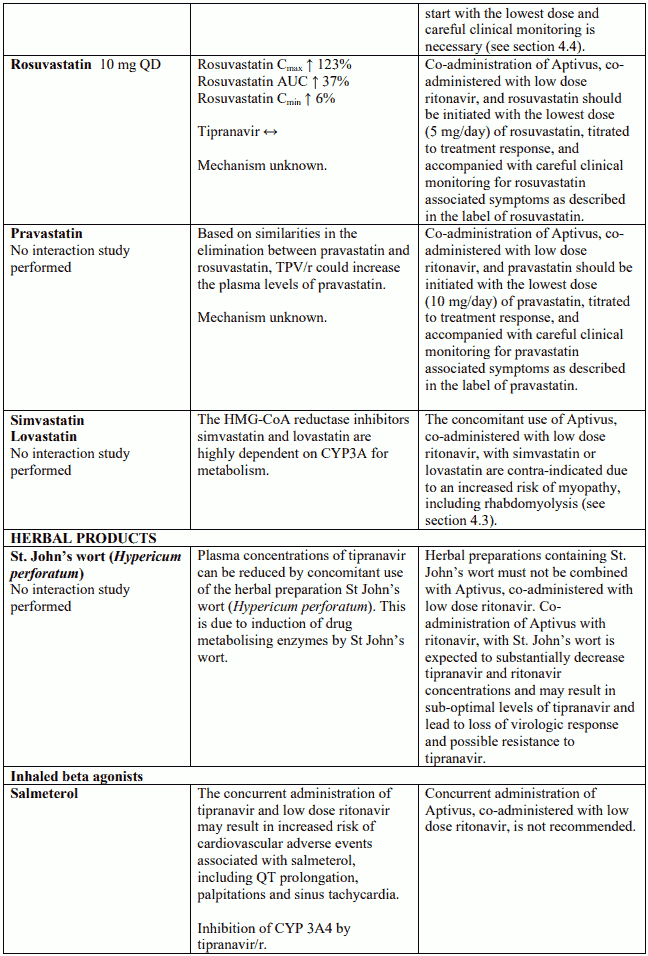
Atorvastatin
Tipranavir, co-administered with low dose ritonavir, increases the plasma concentrations of atorvastatin (see section 4.5). The combination is not recommended. Other HMG-CoA reductase inhibitors should be considered such as pravastatin, fluvastatin or rosuvastatin (see section 4.5). However, if atorvastatin is specifically required for patient management, it should be started with the lowest dose and careful monitoring is necessary.
Omeprazole and other proton pump inhibitors
The combined use of Aptivus with ritonavir with either omeprazole, esomeprazole or with other proton pump inhibitors is not recommended (see section 4.5).
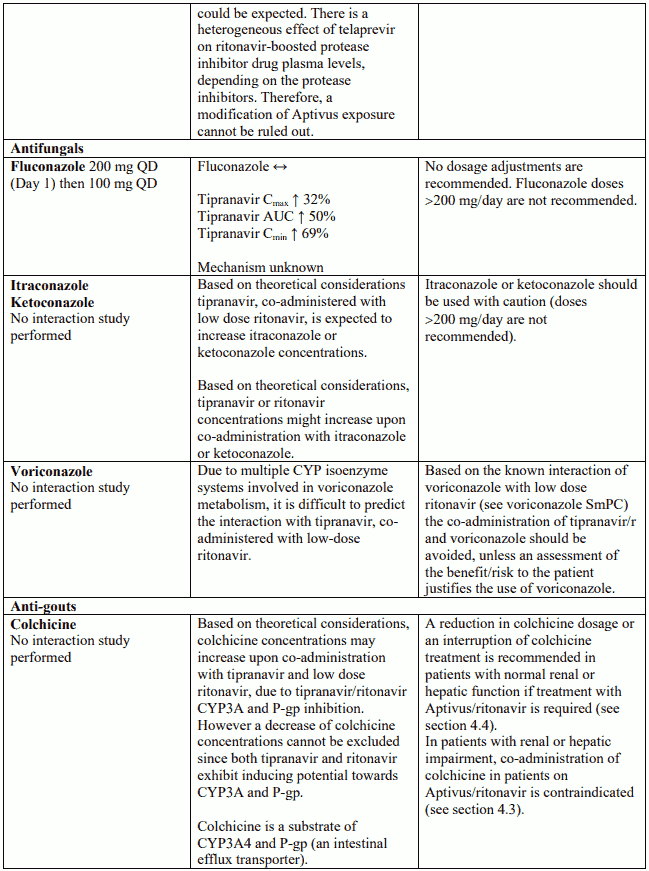
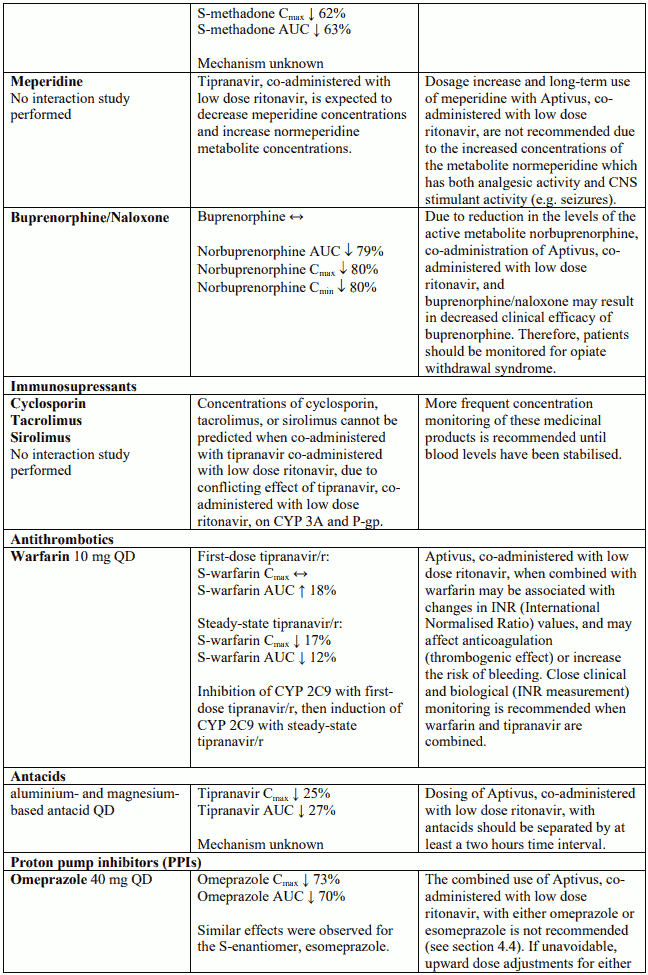
Colchicine
In patients with normal renal and hepatic function, a reduction in colchicine dosage or an interruption of colchicine treatment is recommended in co-administration (see section 4.5).
Salmeterol
Concomitant use of salmeterol and Aptivus, co-administered with low dose ritonavir, is not recommended (see section 4.5).
Bosentan
Due to the marked hepatotoxicity of bosentan and the potential for increasing the liver toxicity associated with Aptivus, co-administered with low dose ritonavir, this combination is not recommended.
Warnings related to certain excipients
Due to Aptivus containing small amounts of sorbitol, patients with rare hereditary problems of fructose intolerance should not take this medicine.
Aptivus contains macrogolglycerol ricinoleate which may cause stomach upset and diarrhoea.
This medicinal product contains 7 vol % ethanol (alcohol), i.e. up to 400 mg per daily dose, equivalent to 8 ml of beer, or less than 4 ml of wine. Harmful for those suffering from alcoholism. To be taken into account in pregnant or breast-feeding women, children and high-risk groups such as patients with liver disease, or epilepsy.
Interaction with other medicinal products and other forms of interaction
The interaction profile of Aptivus, co-administered with low dose ritonavir, is complex and requires special attention in particular in combination with other antiretroviral agents.
Interaction studies have only been performed in adults.
Metabolic profile of tipranavir
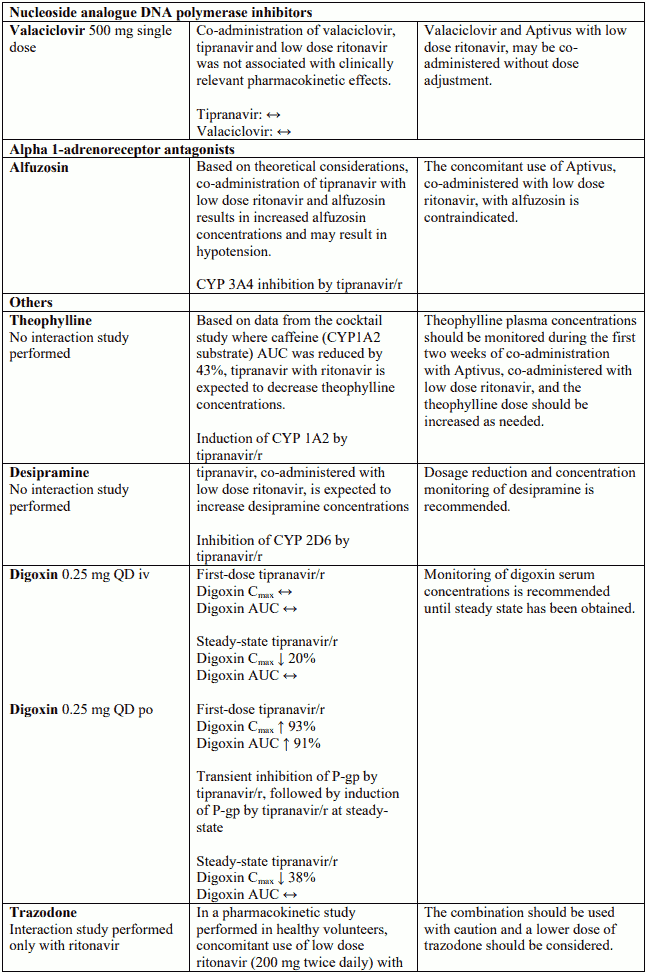
Tipranavir is a substrate, an inducer and an inhibitor of cytochrome P450 CYP3A. When coadministered with ritonavir at the recommended dosage (see section 4.2) there is a net inhibition of P450 CYP3A. Co-administration of Aptivus and low dose ritonavir with agents primarily metabolised by CYP3A may result in changed plasma concentrations of tipranavir or the other agents, which could alter their therapeutic and undesirable effects (see list and details of considered agents, below). Agents that are contraindicated specifically due to the expected magnitude of interaction and potential for serious adverse reactions are detailed in this section, and listed in section 4.3.
A cocktail study was conducted in 16 healthy volunteers with twice-daily 500 mg tipranavir with 200 mg ritonavir capsule administration for 10 days to assess the net effect on the activity of hepatic CYP 1A2 (caffeine), 2C9 (warfarin), 2D6 (dextromethorphan), both intestinal/hepatic CYP 3A4 (midazolam) and P-glycoprotein (P-gp) (digoxin). At steady state, there was a significant induction of CYP 1A2 and a slight induction on CYP 2C9. Potent inhibition of CYP 2D6 and both hepatic and intestinal CYP 3A4 activities were observed. P-gp activity is significantly inhibited after the first dose, but there was a slight induction at steady state. Practical recommendations deriving from this study are displayed below.
Studies in human liver microsomes indicated tipranavir is an inhibitor of CYP 1A2, CYP 2C9, CYP 2C19 and CYP 2D6. The potential net effect of tipranavir with ritonavir on CYP 2D6 is inhibition, because ritonavir is also a CYP 2D6 inhibitor. The in vivo net effect of tipranavir with ritonavir on CYP 1A2, CYP 2C9 and CYP 2C19, indicates, through a preliminary study, an inducing potential of tipranavir with ritonavir on CYP1A2 and, to a lesser extent, on CYP2C9 and P-gp after several days of treatment. Data are not available to indicate whether tipranavir inhibits or induces glucuronosyl transferases.
In vitro studies show that tipranavir is a substrate and also an inhibitor of P-gp.
It is difficult to predict the net effect of Aptivus co-administered with low dose ritonavir on oral bioavailability and plasma concentrations of agents that are dual substrates of CYP3A and P-gp. The net effect will vary depending on the relative affinity of the co-administered substance for CYP3A and P-gp, and the extent of intestinal first-pass metabolism/efflux.
Co-administration of Aptivus and agents that induce CYP3A and/or P-gp may decrease tipranavir concentrations and reduce its therapeutic effect (see list and details of considered agents, below). Coadministration of Aptivus and medicinal products that inhibit P-gp may increase tipranavir plasma concentrations.
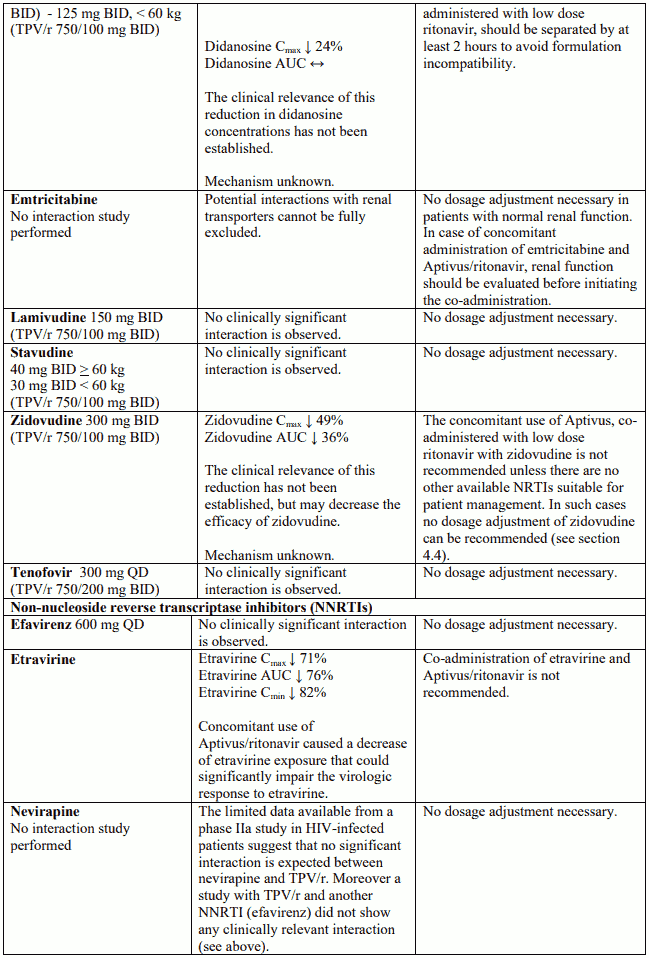
Known and theoretical interactions with selected antiretrovirals and non-antiretroviral medicinal products are listed in the table below.
Interaction table
Interactions between Aptivus and co-administered medicinal products are listed in the table below (increase is indicated as "↑", decrease as "↓", no change as "↔",once daily as "QD", twice daily as "BID"). Unless otherwise stated, studies detailed below have been performed with the recommended dosage of Aptivus/r (i.e. 500/200 mg BID). However, some PK interaction studies were not performed with this recommended dosage. Nevertheless, the results of many of these interaction studies can be extrapolated to the recommended dosage since the doses used (eg. TPV/r 500/100 mg, TPV/r 750/200 mg) represented extremes of hepatic enzyme induction and inhibition and bracketed the recommended dosage of Aptivus/r.


Pregnancy and lactation
Contraception in males and females
Tipranavir adversely interacts with oral contraceptives. Therefore, an alternative, effective, safe method of contraception should be used during treatment (see section 4.5).
Pregnancy
There are no adequate data from the use of tipranavir in pregnant women. Studies in animals have shown reproductive toxicity (see section 5.3). The potential risk for humans is unknown. Tipranavir should be used during pregnancy only if the potential benefit justifies the potential risk to the foetus.
Breastfeeding
Consistent with the recommendation that HIV-infected mothers should not breast-feed their infants under any circumstances to avoid risking postnatal transmission of HIV, mothers should discontinue breast-feeding if they are receiving Aptivus.
Fertility
Clinical data on fertility are not available for tipranavir. Preclinical studies performed with tipranavir showed no adverse effect on fertility (see section 5.3).
Effects on ability to drive and use machines
Dizziness, somnolence, and fatigue have been reported in some patients; therefore, caution should be recommended when driving a car or operating machinery. If patients experience fatigue, dizziness, or somnolence they should avoid potentially hazardous tasks such as driving or operating machinery.
Undesirable effects
Summary of the safety profile
Amongst the most common adverse reactions reported for Aptivus were gastrointestinal complaints such as diarrhoea and nausea as well as hyperlipidaemia. The most serious adverse reactions include hepatic impairment and liver toxicity. Intracranial haemorrhage (ICH) was only observed in post marketing experience (see section 4.4).
Aptivus co-administered with low dose ritonavir, has been associated with reports of significant liver toxicity. In Phase III RESIST trials, the frequency of transaminase elevations was significantly increased in the tipranavir with ritonavir arm compared to the comparator arm. Close monitoring is therefore needed in patients treated with Aptivus, co-administered with low dose ritonavir (see section 4.4).
Limited data are currently available for the use of Aptivus, co-administered with low dose ritonavir, in patients co-infected with hepatitis B or C. Aptivus should therefore be used with caution in patients co-infected with hepatitis B or C. Aptivus should be used in this patient population only if the potential benefit outweighs the potential risk, and with increased clinical and laboratory monitoring.
Tabulated summary of adverse reactions
Assessment of adverse reactions from HIV-1 clinical study data is based on experience in all Phase II and III trials in adults treated with the 500 mg tipranavir with 200 mg ritonavir dose twice daily (n=1397) and are listed below by system organ class and frequency according to the following categories: Very common (≥1/10), common (≥1/100 to 1/10), uncommon (≥1/1,000 to 1/100), rare (≥1/10,000 to 1/1,000).
Tabulated summary of adverse reactions associated with Aptivus based on clinical studies and postmarketing experience:
Blood and lymphatic system disorders
Uncommon: neutropenia, anaemia, thrombocytopenia
Immune system disorders
Uncommon: hypersensitivity
Metabolism and nutrition disorders
Common: hypertriglyceridaemia, hyperlipidaemia
Uncommon: anorexia, decreased appetite, weight decreased, hyperamylasaemia, hypercholesterolaemia, diabetes mellitus, hyperglycaemia
Rare: dehydration
Psychiatric disorders
Uncommon: insomnia, sleep disorder
Nervous system disorders
Common: headache
Uncommon: dizziness, neuropathy peripheral, somnolence
Rare: intracranial haemorrhage*
Respiratory, thoracic and mediastinal disorders
Uncommon: dyspnoea
Gastrointestinal disorders
Very common: diarrhoea, nausea
Common: vomiting, flatulence, abdominal pain, abdominal distension, dyspepsia
Uncommon: gastrooesophageal reflux disease, pancreatitis
Rare: lipase increased
Hepatobiliary disorders
Uncommon: hepatic enzyme increased (ALAT, ASAT), cytolytic hepatitis, liver function test abnormal (ALAT, ASAT), hepatitis toxic
Rare: hepatic failure (including fatal outcome), hepatitis, hepatic steatosis, hyperbilirubinaemia
Skin and subcutaneous tissue disorders
Common: rash
Uncommon: pruritus, exanthem
Musculoskeletal and connective tissue disorders
Uncommon: myalgia, muscle spasms
Renal and urinary disorders
Uncommon: renal failure
General disorders and administration site conditions
Common: fatigue
Uncomm: pyrexia, influenza like illness, malaise
* see section Description of selected adverse reactions "Bleeding" for source of information
Description of selected adverse reactions
The following clinical safety features (hepatotoxicity, hyperlipidaemia, bleeding events, rash) were seen at higher frequency among tipranavir with ritonavir treated patients when compared with the comparator arm treated patients in the RESIST trials, or have been observed with tipranavir with ritonavir administration. The clinical significance of these observations has not been fully explored.
Hepatotoxicity
After 48 weeks of follow-up, the frequency of Grade 3 or 4 ALAT and/or ASAT abnormalities was higher in tipranavir with ritonavir patients compared with comparator arm patients (10% and 3.4%, respectively). Multivariate analyses showed that baseline ALAT or ASAT above DAIDS Grade 1 and co-infection with hepatitis B or C were risk factors for these elevations. Most patients were able to continue treatment with tipranavir with ritonavir.
Metabolic parameters
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4)
Hyperlipidaemia
Grade 3 or 4 elevations of triglycerides occurred more frequently in the tipranavir with ritonavir arm compared with the comparator arm. At 48 weeks these rates were 25.2% of patients in the tipranavir with ritonavir arm and 15.6% in the comparator arm.
Bleeding
This adverse reaction was identified through post-marketing surveillance but not observed in randomised controlled clinical trials (n=6300). RESIST participants receiving tipranavir with ritonavir tended to have an increased risk of bleeding; at 24 weeks the relative risk was 1.98 (95% CI=1.03, 3.80). At 48-weeks the relative risk decreased to 1.27 (95% CI=0.76, 2.12). There was no pattern for the bleeding events and no difference between treatment groups in coagulation parameters. The significance of this finding is being further monitored.
Fatal and non-fatal intracranial haemorrhage (ICH) have been reported in patients receiving tipranavir, many of whom had other medical conditions or were receiving concomitant medicinal products that may have caused or contributed to these events. However, in some cases the role of tipranavir cannot be excluded. No pattern of abnormal haematological or coagulation parameters has been observed in patients in general, or preceding the development of ICH. Therefore, routine measurement of coagulation parameters is not currently indicated in the management of patients on Aptivus. An increased risk of ICH has previously been observed in patients with advanced HIV disease/AIDS such as those treated in the Aptivus clinical trials.
Rash
An interaction study in women between tipranavir, co-administered with low dose ritonavir, and ethinyl oestradiol/norethindrone demonstrated a high frequency of non-serious rash. In the RESIST trials, the risk of rash was similar between tipranavir with ritonavir and comparator arms (16.3% vs. 12.5%, respectively; see section 4.4). No cases of Stevens-Johnson Syndrome or Toxic Epidermal Necrolysis have been reported in the clinical development programme of tipranavir.
Laboratory abnormalities
Frequencies of marked clinical laboratory abnormalities (Grade 3 or 4) reported in at least 2% of patients in the tipranavir with ritonavir arms in the phase III clinical studies (RESIST-1 and RESIST2) after 48-weeks were increased ASAT (6.1%), increased ALAT (9.7%), increased amylase (6.0%), increased cholesterol (4.2%), increased triglycerides (24.9%), and decreased white blood cell count (5.7%).
Increased CPK, myalgia, myositis and, rarely, rhabdomyolysis, have been reported with protease inhibitors, particularly in combination with nucleoside reverse transcriptase inhibitors.
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4). Reactivation of herpes simplex and herpes zoster virus infections were observed in the RESIST trials.
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
Paediatric population
In an open-label, dose-finding study of tipranavir plus ritonavir (Trial 1182.14), 28 children who were 12 years of age or above received Aptivus capsules. In general, adverse reactions were similar to those seen in adults, with the exception of vomiting, rash and pyrexia, which were reported more frequently in children than in adults. The most frequently reported moderate or severe adverse reactions in the 48 week analyses are noted below.
Most frequently reported moderate or severe adverse reactions in paediatric patients aged 12 to 18 years who took Aptivus capsules (reported in 2 or more children, Trial 1182.14, week 48 analyses, Full Analysis Set).
| Total patients treated (N) | 28 |
|---|---|
| Events [N(%)] | |
| Vomiting/ retching | 3 (10.7) |
| Nausea | 2 (7.1) |
| Abdominal pain1 | 2 (7.1) |
| Rash2 | 3 (10.7) |
| Insomnia | 2 (7.1) |
| ALAT increased | 4 (14.3) |
1 Includes abdominal pain (N=1) and dyspepsia (N=1).
2 Rash consists of one or more of the preferred terms of rash, drug eruption, rash macular, rash papular, erythema, rash maculo-papular, rash pruritic, and urticaria
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.