ARIMIDEX Film-coated tablets Ref.[6190] Active ingredients: Anastrozole
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2017 Publisher: AstraZeneca UK Limited, 600 Capability Green, Luton, LU1 3LU, UK
Pharmacodynamic properties
Pharmacotherapeutic group: Enzyme inhibitors
ATC code: L02BG03
Mechanism of action and pharmacodynamic effects
Arimidex is a potent and highly selective non-steroidal aromatase inhibitor. In postmenopausal women, estradiol is produced primarily from the conversion of androstenedione to estrone through the aromatase enzyme complex in peripheral tissues. Estrone is subsequently converted to estradiol. Reducing circulating estradiol levels has been shown to produce a beneficial effect in women with breast cancer. In postmenopausal women, Arimidex at a daily dose of 1 mg produced estradiol suppression of greater than 80% using a highly sensitive assay.
Arimidex does not possess any progestogenic, androgenic, or estrogenic activity.
Daily doses of Arimidex up to 10 mg do not have any effect on cortisol or aldosterone secretion, measured before or after standard adrenocorticotrophic hormone (ACTH) challenge testing. Corticoid supplements are therefore not needed.
Clinical efficacy and safety
Advanced breast cancer
First-line therapy in postmenopausal women with advanced breast cancer
Two double-blind, controlled clinical studies of similar design (Study 1033IL/0030 and Study 1033IL/0027) were conducted to assess the efficacy of Arimidex compared with tamoxifen as first-line therapy for hormone receptor-positive or hormone receptor-unknown locally advanced or metastatic breast cancer in postmenopausal women. A total of 1,021 patients were randomised to receive 1 mg of Arimidex once daily or 20 mg of tamoxifen once daily. The primary endpoints for both trials were time to tumour progression, objective tumour response rate, and safety.
For the primary endpoints, Study 1033IL/0030 showed that Arimidex had a statistically significant advantage over tamoxifen for time to tumour progression (Hazard ratio (HR) 1.42, 95% Confidence Interval (CI) [1.11, 1.82], Median time to progression 11.1 and 5.6 months for Arimidex and tamoxifen respectively, p=0.006); objective tumour response rates were similar for Arimidex and tamoxifen. Study 1033IL/0027 showed that Arimidex and tamoxifen had similar objective tumour response rates and time to tumour progression. Results from the secondary endpoints were supportive of the results of the primary efficacy endpoints. There were too few deaths occurring across treatment groups of both trials to draw conclusions on overall survival differences.
Second-line therapy in postmenopausal women with advanced breast cancer
Arimidex was studied in two controlled clinical trials (Study 0004 and Study 0005) in postmenopausal women with advanced breast cancer who had disease progression following tamoxifen therapy for either advanced or early breast cancer. A total of 764 patients were randomised to receive either a single daily dose of 1 mg or 10 mg of Arimidex or megestrol acetate 40 mg four times a day. Time to progression and objective response rates were the primary efficacy variables. The rate of prolonged (more than 24 weeks) stable disease, the rate of progression, and survival were also calculated. In both studies there were no significant differences between treatment arms with respect to any of the efficacy parameters.
Adjuvant treatment of early invasive breast cancer for hormone receptor-positive patients
In a large phase III study conducted in 9,366 postmenopausal women with operable breast cancer treated for 5 years (see below), Arimidex was shown to be statistically superior to tamoxifen in disease-free survival. A greater magnitude of benefit was observed for disease-free survival in favour of Arimidex versus tamoxifen for the prospectively defined hormone receptor-positive population.
Table 3. ATAC endpoint summary: 5-year treatment completion analysis:
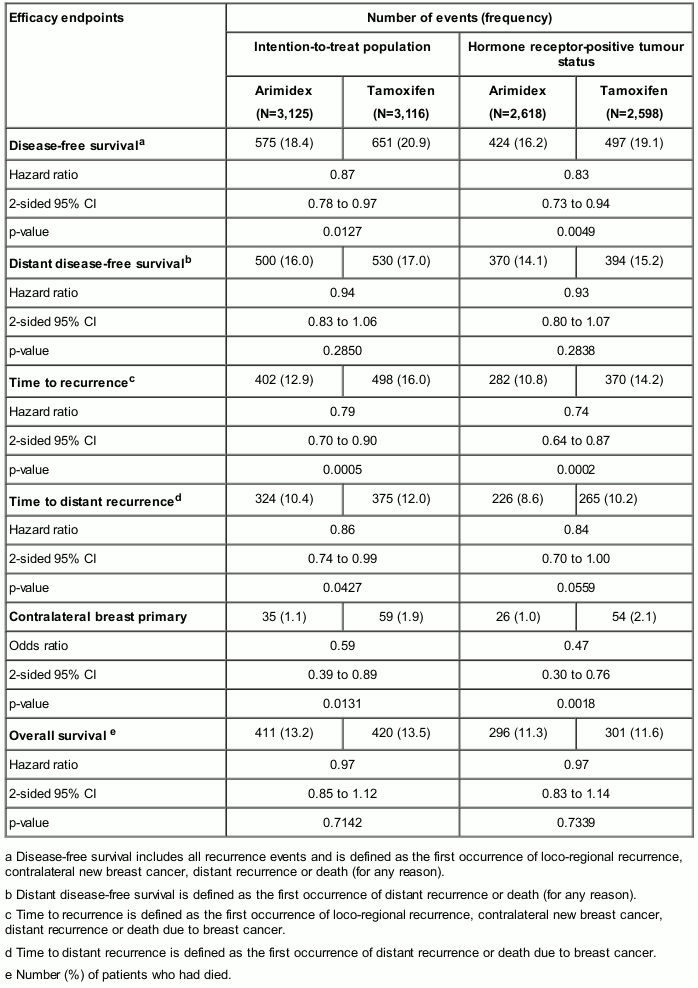
The combination of Arimidex and tamoxifen did not demonstrate any efficacy benefits in comparison with tamoxifen in all patients as well as in the hormone receptor-positive population. This treatment arm was discontinued from the study.
With an updated follow-up at a median of 10 years, long-term comparison of the treatment effects of Arimidex relative to tamoxifen were shown to be consistent with previous analyses.
Adjuvant treatment of early invasive breast cancer for hormone receptor-positive patients being treated with adjuvant tamoxifen
In a phase III trial (Austrian Breast and Colorectal Cancer Study Group [ABCSG] 8) conducted in 2,579 postmenopausal women with hormone receptor-positive early breast cancer who had received surgery with or without radiotherapy and no chemotherapy (see below), switching to Arimidex after 2 years adjuvant treatment with tamoxifen was statistically superior in disease-free survival when compared to remaining on tamoxifen, after a median follow-up of 24 months.
Table 4. ABCSG 8 trial endpoint and results summary:
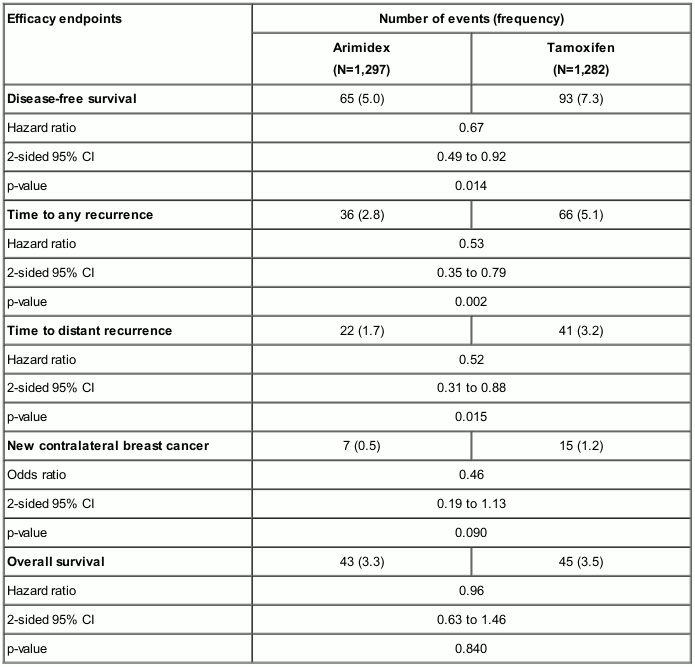
Two further similar trials (GABG/ARNO 95 and ITA), in one of which patients had received surgery and chemotherapy, as well as a combined analysis of ABCSG 8 and GABG/ARNO 95, supported these results.
The Arimidex safety profile in these 3 studies was consistent with the known safety profile established in postmenopausal women with hormone receptor-positive early breast cancer.
Bone mineral density (BMD)
In the phase III/IV study (Study of Anastrozole with the Bisphosphonate Risedronate [SABRE]), 234 postmenopausal women with hormone receptor-positive early breast cancer scheduled for treatment with Arimidex 1 mg/day were stratified to low, moderate and high risk groups according to their existing risk of fragility fracture. The primary efficacy parameter was the analysis of lumbar spine bone mass density using DEXA scanning. All patients received treatment with vitamin D and calcium. Patients in the low risk group received Arimidex alone (N=42), those in the moderate group were randomised to Arimidex plus risedronate 35 mg once a week (N=77) or Arimidex plus placebo (N=77) and those in the high risk group received Arimidex plus risedronate 35 mg once a week (N=38). The primary endpoint was change from baseline in lumbar spine bone mass density at 12 months.
The 12-month main analysis has shown that patients already at moderate to high risk of fragility fracture showed no decrease in their bone mass density (assessed by lumbar spine bone mineral density using DEXA scanning) when managed by using Arimidex 1 mg/day in combination with risedronate 35 mg once a week. In addition, a decrease in BMD which was not statistically significant was seen in the low risk group treated with Arimidex 1 mg/day alone. These findings were mirrored in the secondary efficacy variable of change from baseline in total hip BMD at 12 months.
This study provides evidence that the use of bisphosphonates could be considered in the management of possible bone mineral loss in postmenopausal women with early breast cancer scheduled to be treated with Arimidex.
Paediatric population
Arimidex is not indicated for use in children and adolescents. Efficacy has not been established in the paediatric populations studied (see below). The number of children treated was too limited to draw any reliable conclusions on safety. No data on the potential long-term effects of Arimidex treatment in children and adolescents are available (see section 5.3).
The European Medicines Agency has waived the obligation to submit the results of studies with Arimidex in one or several subsets of the paediatric population in short stature due to growth hormone deficiency (GHD), testotoxicosis, gynaecomastia, and McCune-Albright syndrome (see section 4.2).
Short stature due to Growth Hormone Deficiency
A randomised, double-blind, multi-centre study evaluated 52 pubertal boys (aged 11 to 16 years inclusive) with GHD treated for 12 to 36 months with Arimidex 1 mg/day or placebo in combination with growth hormone. Only 14 subjects on Arimidex completed 36 months.
No statistically significant difference from placebo was observed for the growth related parameters of predicted adult height, height, height SDS (standard deviation score), and height velocity. Final height data were not available. While the number of children treated was too limited to draw any reliable conclusions on safety, there was an increased fracture rate and a trend towards reduced bone mineral density in the Arimidex arm compared to placebo.
Testotoxicosis
An open-label, non-comparative, multi-centre study evaluated 14 male patients (aged 2 to 9 years) with familial male-limited precocious puberty, also known as testotoxicosis, treated with combination of Arimidex and bicalutamide. The primary objective was to assess the efficacy and safety of this combination regimen over 12 months. Thirteen out of the 14 patients enrolled completed 12 months of combination treatment (one patient was lost to follow-up). There was no significant difference in growth rate after 12 months of treatment, relative to the growth rate during the 6 months prior to entering the study.
Gynaecomastia studies
Trial 0006 was a randomised, double-blind, multi-centre study of 82 pubertal boys (aged 11-18 years inclusive) with gynaecomastia of greater than 12 months duration treated with Arimidex 1 mg/day or placebo daily for up to 6 months. No significant difference in the number of patients who had a 50% or greater reduction in total breast volume after 6 months of treatment was observed between the Arimidex 1 mg treated group and the placebo group.
Trial 0001 was an open-label, multiple-dose pharmacokinetic study of Arimidex 1 mg/day in 36 pubertal boys with gynaecomastia of less than 12 months duration. The secondary objectives were to evaluate the proportion of patients with reductions from baseline in the calculated volume of gynaecomastia of both breasts combined of at least 50% between day 1 and after 6 months of study treatment, and patient tolerability and safety. A decrease in 50% or more of total breast volume was seen in 56% (20/36) of the boys after 6 months.
McCune-Albright Syndrome study
Trial 0046 was an international, multi-centre, open-label exploratory trial of Arimidex in 28 girls (aged 2 to ≤10 years) with McCune-Albright Syndrome (MAS). The primary objective was to evaluate the safety and efficacy of Arimidex 1 mg/day in patients with MAS. The efficacy of study treatment was based on the proportion of patients fulfilling defined criteria relating to vaginal bleeding, bone age, and growth velocity.
No statistically significant change in the frequency of vaginal bleeding days on treatment was observed. There were no clinically significant changes in Tanner staging, mean ovarian volume, or mean uterine volume. No statistically significant change in the rate of increase in bone age on treatment compared to the rate during baseline was observed. Growth rate (in cm/year) was significantly reduced (p<0.05) from pre-treatment through month 0 to month 12, and from pre-treatment to the second 6 months (month 7 to month 12).
Pharmacokinetic properties
Absorption
Absorption of anastrozole is rapid and maximum plasma concentrations typically occur within two hours of dosing (under fasted conditions). Food slightly decreases the rate but not the extent of absorption. The small change in the rate of absorption is not expected to result in a clinically significant effect on steady-state plasma concentrations during once daily dosing of Arimidex tablets. Approximately 90 to 95% of plasma anastrozole steady-state concentrations are attained after 7 daily doses, and accumulation is 3- to 4-fold. There is no evidence of time or dose-dependency of anastrozole pharmacokinetic parameters.
Anastrozole pharmacokinetics are independent of age in postmenopausal women.
Distribution
Anastrozole is only 40% bound to plasma proteins.
Elimination
Anastrozole is eliminated slowly with a plasma elimination half-life of 40 to 50 hours. Anastrozole is extensively metabolised by postmenopausal women with less than 10% of the dose excreted in the urine unchanged within 72 hours of dosing. Metabolism of anastrozole occurs by N-dealkylation, hydroxylation and glucuronidation. The metabolites are excreted primarily via the urine. Triazole, the major metabolite in plasma, does not inhibit aromatase.
Renal or hepatic impairment
The apparent clearance (CL/F) of anastrozole, following oral administration, was approximately 30% lower in volunteers with stable hepatic cirrhosis than in matched controls (Study 1033IL/0014). However, plasma anastrozole concentrations in the volunteers with hepatic cirrhosis were within the range of concentrations seen in normal subjects in other trials. Plasma anastrozole concentrations observed during long-term efficacy trials in patients with hepatic impairment were within the range of plasma anastrozole concentrations seen in patients without hepatic impairment.
The apparent clearance (CL/F) of anastrozole, following oral administration, was not altered in volunteers with severe renal impairment (GFR <30ml/min) in Study 1033IL/0018, consistent with the fact that anastrozole is eliminated primarily by metabolism. Plasma anastrozole concentrations observed during long-term efficacy trials in patients with renal impairment were within the range of plasma anastrozole concentrations seen in patients without renal impairment. In patients with severe renal impairment, administration of Arimidex should be performed with caution (see section 4.2 and 4.4).
Paediatric population
In boys with pubertal gynaecomastia (10-17 years), anastrozole was rapidly absorbed, was widely distributed, and was eliminated slowly with a half-life of approximately 2 days. Clearance of anastrozole was lower in girls (3-10 years) than in the older boys and exposure higher. Anastrozole in girls was widely distributed and slowly eliminated.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction for the indicated population.
Acute toxicity
In animal studies toxicity was only seen at high doses. In acute toxicity studies in rodents, the median lethal dose of anastrozole was greater than 100 mg/kg/day by the oral route and greater than 50 mg/kg/day by the intraperitoneal route. In an oral acute toxicity study in the dog, the median lethal dose was greater than 45 mg/kg/day.
Chronic toxicity
In animal studies adverse effects were only seen at high doses. Multiple dose toxicity studies utilised rats and dogs. No no-effect levels were established for anastrozole in the toxicity studies, but those effects that were observed at the low doses (1 mg/kg/day) and mid doses (dog 3 mg/kg/day; rat 5 mg/kg/day) were related to either the pharmacological or enzyme inducing properties of anastrozole and were unaccompanied by significant toxic or degenerative changes.
Mutagenicity
Genetic toxicology studies with anastrozole show that it is not a mutagen or a clastogen.
Reproductive toxicology
In a fertility study weanling male rats were dosed orally with 50 or 400 mg/l anastrozole via their drinking water for 10 weeks. Measured mean plasma concentrations were 44.4 (±14.7) ng/ml and 165 (±90) ng/ml respectively. Mating indices were adversely affected in both dose groups, whilst a reduction in fertility was evident only at the 400 mg/l dose level. The reduction was transient as all mating and fertility parameters were similar to control group values following a 9-week treatment-free recovery period.
Oral administration of anastrozole to female rats produced a high incidence of infertility at 1 mg/kg/day and increased pre-implantation loss at 0.02 mg/kg/day. These effects occurred at clinically relevant doses. An effect in man cannot be excluded. These effects were related to the pharmacology of the compound and were completely reversed after a 5-week compound withdrawal period.
Oral administration of anastrozole to pregnant rats and rabbits caused no teratogenic effects at doses up to 1.0 and 0.2 mg/kg/day respectively. Those effects that were seen (placental enlargement in rats and pregnancy failure in rabbits) were related to the pharmacology of the compound.
The survival of litters born to rats given anastrozole at 0.02 mg/kg/day and above (from Day 17 of pregnancy to Day 22 post-partum) was compromised. These effects were related to the pharmacological effects of the compound on parturition. There were no adverse effects on behaviour or reproductive performance of the first generation offspring attributable to maternal treatment with anastrozole.
Carcinogenicity
A two-year rat oncogenicity study resulted in an increase in incidence of hepatic neoplasms and uterine stromal polyps in females and thyroid adenomas in males at the high dose (25 mg/kg/day) only. These changes occurred at a dose which represents 100-fold greater exposure than occurs at human therapeutic doses, and are considered not to be clinically relevant to the treatment of patients with anastrozole.
A two-year mouse oncogenicity study resulted in the induction of benign ovarian tumours and a disturbance in the incidence of lymphoreticular neoplasms (fewer histiocytic sarcomas in females and more deaths as a result of lymphomas). These changes are considered to be mouse-specific effects of aromatase inhibition and not clinically relevant to the treatment of patients with anastrozole.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.