BAFIERTAM Capsule Ref.[10345] Active ingredients:
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
The mechanism by which monomethyl fumarate (MMF) exerts its therapeutic effect in multiple sclerosis is unknown. MMF has been shown to activate the Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) pathway in vitro and in vivo in animals and humans. The Nrf2 pathway is involved in the cellular response to oxidative stress. MMF has been identified as a nicotinic acid receptor agonist in vitro.
12.2. Pharmacodynamics
Potential to prolong the QT interval
In a placebo controlled thorough QT study performed in healthy subjects with dimethyl fumarate [the prodrug of BAFIERTAM], there was no evidence that dimethyl fumarate caused QT interval prolongation of clinical significance (i.e., the upper bound of the 90% confidence interval for the largest placebo-adjusted, baseline-corrected QTc was below 10 ms).
12.3. Pharmacokinetics
Pharmacokinetics of monomethyl fumarate have previously been characterized after oral administration of its prodrug, dimethyl fumarate, as delayed-release capsules, in healthy subjects and subjects with multiple sclerosis. After oral administration, dimethyl fumarate undergoes rapid presystemic hydrolysis by esterases and is converted to its active metabolite, monomethyl fumarate (MMF). Additional pharmacokinetic data of monomethyl fumarate were obtained after oral administration of BAFIERTAM, the monomethyl fumarate delayed-release capsules, in healthy subjects.
Absorption
Following oral administration of BAFIERTAM 190 mg (two 95 mg monomethyl fumarate delayedrelease capsules) under fasting conditions, the median Tmax of MMF is 4.03 hours; and the peak plasma concentration (Cmax) and overall exposure (AUC) of monomethyl fumarate are bioequivalent to those after oral administration of 240 mg dimethyl fumarate delayed-release capsule. A high-fat, high-calorie meal did not significantly affect the overall monomethyl fumarate plasma exposure (AUC), but decreased the Cmax of MMF by 20%, with prolonged absorption. The median Tmax of MMF was delayed from approximately 4.0 hours to 11 hours by a high fat meal.
Distribution
From studies with dimethyl fumarate (the prodrug of BAFIERTAM), it is shown that the apparent volume of distribution of MMF varies between 53 and 73 L in healthy subjects. Human plasma protein binding of MMF is 27-45% and independent of concentration.
Elimination
Metabolism
In humans, metabolism of MMF occurs through the tricarboxylic acid (TCA) cycle, with no involvement of the cytochrome P450 (CYP450) system. MMF, fumaric and citric acid, and glucose are the major metabolites of MMF in plasma.
Excretion
From studies with dimethyl fumarate (the prodrug of BAFIERTAM), exhalation of CO2 is the primary route of elimination, accounting for approximately 60% of the dimethyl fumarate dose. Renal and fecal elimination are minor routes of elimination for MMF, accounting for 16% and 1% of the dimethyl fumarate dose respectively. Trace amounts of unchanged MMF were present in urine.
The plasma half-life of MMF is approximately 0.5 hour and no circulating MMF is present at 24 hours in the majority of individual following oral administration of BAFIERTAM 190 mg (two 95 mg monomethyl fumarate delayed-release capsules) under fasting conditions. Accumulation of MMF does not occur with multiple doses of dimethyl fumarate.
Specific Populations
Body weight, gender, and age differences do not require dosage adjustment.
No studies have been conducted in subjects with hepatic or renal impairment. However, neither condition would be expected to affect plasma exposure to MMF and therefore no dosage adjustment is necessary.
Drug Interaction Studies
No potential drug interactions with dimethyl fumarate or MMF were identified in in vitro CYP inhibition and induction studies, or in P-glycoprotein studies. Single doses of interferon beta-1a or glatiramer acetate did not alter the pharmacokinetics of MMF. Aspirin, when administered approximately 30 minutes before dimethyl fumarate, did not alter the pharmacokinetics of MMF.
Oral Contraceptives
The coadministration of the prodrug of BAFIERTAM, dimethyl fumarate, with a combined oral contraceptive (norelgestromin and ethinyl estradiol) did not elicit any relevant effects in oral contraceptives exposure. No interaction studies have been performed with oral contraceptives containing other progestogens.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies of dimethyl fumarate (DMF) were conducted in mice and rats. In mice, oral administration of DMF (0, 25, 75, 200, and 400 mg/kg/day) for up to two years resulted in an increase in nonglandular stomach (forestomach) and kidney tumors: squamous cell carcinomas and papillomas of the forestomach in males and females at 200 and 400 mg/kg/day; leiomyosarcomas of the forestomach at 400 mg/kg/day in males and females; renal tubular adenomas and carcinomas at 200 and 400 mg/kg/day in males; and renal tubule adenomas at 400 mg/kg/day in females. Plasma MMF exposure (AUC) at the highest dose not associated with tumors in mice (75 mg/kg/day) was similar to that in humans at the recommended human dose (RHD) of MMF (380 mg/day).
In rats, oral administration of DMF (0, 25, 50, 100, and 150 mg/kg/day) for up to two years resulted in increases in squamous cell carcinomas and papillomas of the forestomach at all doses tested in males and females, and in testicular interstitial (Leydig) cell adenomas at 100 and 150 mg/kg/day. Plasma MMF AUC at the lowest dose tested was lower than that in humans at the RHD of MMF.
Mutagenesis
Monomethyl fumarate (MMF) was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. MMF was clastogenic in the in vitro chromosomal aberration assay in human peripheral blood lymphocytes in the absence of metabolic activation. DMF was not clastogenic in the in vivo micronucleus assay in the rat.
Impairment of Fertility
In male rats, oral administration of DMF (0, 75, 250, and 375 mg/kg/day) prior to and throughout the mating period had no effect on fertility; however, increases in non-motile sperm were observed at the mid and high doses. The no-effect dose for adverse effects on sperm is similar to the recommended human dose (RHD) of DMF (480 mg/day) on a body surface area (mg/m2) basis. MMF (380 mg/day) is bioequivalent to DMF (480 mg/day).
In female rats, oral administration of DMF (0, 20, 100, and 250 mg/kg/day) prior to and during mating and continuing to gestation day 7 caused disruption of the estrous cycle and increases in embryolethality at the highest dose tested. The highest dose not associated with adverse effects (100 mg/kg/day) is twice the RHD for DMF on a mg/m2 basis. MMF (380 mg/day) is bioequivalent to DMF (480 mg/day).
Testicular toxicity (germinal epithelial degeneration, atrophy, hypospermia, and/or hyperplasia) was observed at clinically relevant doses in mice, rats, and dogs in subchronic and chronic oral toxicity studies of DMF, and in a chronic oral toxicity study evaluating a combination of four fumaric acid esters (including DMF) in rats.
13.2. Animal Toxicology and/or Pharmacology
Kidney toxicity was observed after repeated oral administration of dimethyl fumarate (DMF) in mice, rats, dogs, and monkeys. Renal tubule epithelia regeneration, suggestive of tubule epithelial injury, was observed in all species. Renal tubular hyperplasia was observed in rats with dosing for up to two years. Cortical atrophy and interstitial fibrosis were observed in dogs and monkeys at doses above 5 mg/kg/day. In monkeys, the highest dose tested (75 mg/kg/day) was associated with single cell necrosis and multifocal and diffuse interstitial fibrosis, indicating irreversible loss of renal tissue and function. In dogs and monkeys, the 5 mg/kg/day dose was associated with plasma MMF exposures less than or similar to that in humans at the recommended human dose (RHD) of MMF (380 mg/day).
A dose-related increase in incidence and severity of retinal degeneration was observed in mice following oral administration of DMF for up to two years at doses above 75 mg/kg/day, a dose associated with plasma MMF exposure (AUC) similar to that in humans at the RHD of MMF (380 mg/day).
14. Clinical Studies
The efficacy of BAFIERTAM is based upon bioavailability studies in healthy subjects comparing oral dimethyl fumarate delayed-release capsules to BAFIERTAM delayed-release capsules [see Clinical Pharmacology (12.3)].
The clinical studies described below were conducted using dimethyl fumarate.
The efficacy and safety of dimethyl fumarate were demonstrated in two studies (Studies 1 and 2) that evaluated dimethyl fumarate taken either twice or three times a day in patients with relapsingremitting multiple sclerosis (RRMS). The starting dose for dimethyl fumarate was 120 mg twice or three times a day for the first 7 days, followed by an increase to 240 mg twice or three times a day. Both studies included patients who had experienced at least 1 relapse over the year preceding the trial or had a brain Magnetic Resonance Imaging (MRI) scan demonstrating at least one gadoliniumenhancing (Gd+) lesion within 6 weeks of randomization. The Expanded Disability Status Scale (EDSS) was also assessed and patients could have scores ranging from 0 to 5. Neurological evaluations were performed at baseline, every 3 months, and at the time of suspected relapse. MRI evaluations were performed at baseline, month 6, and year 1 and 2 in a subset of patients (44% in Study 1 and 48% in Study 2).
Study 1. Placebo-Controlled Trial in RRMS
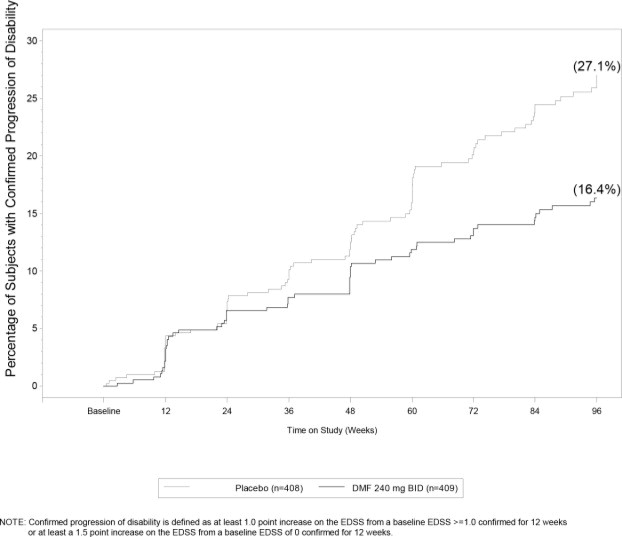
Study 1 was a 2-year randomized, double-blind, placebo-controlled study in 1234 patients with RRMS. The primary endpoint was the proportion of patients relapsed at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of new T1 hypointense lesions, number of Gd+ lesions, annualized relapse rate (ARR), and time to confirmed disability progression. Confirmed disability progression was defined as at least a 1 point increase from baseline EDSS (1.5 point increase for patients with baseline EDSS of 0) sustained for 12 weeks.
Patients were randomized to receive dimethyl fumarate 240 mg twice a day (n=410), dimethyl fumarate 240 mg three times a day (n=416), or placebo (n=408) for up to 2 years. The median age was 39 years, median time since diagnosis was 4 years, and median EDSS score at baseline was 2. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 69% for patients assigned to dimethyl fumarate 240 mg twice a day, 69% for patients assigned to dimethyl fumarate 240 mg three times a day, and 65% for patients assigned to placebo groups.
Dimethyl fumarate had a statistically significant effect on all of the endpoints described above and the 240 mg three times daily dose showed no additional benefit over the dimethyl fumarate 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 2 and Figure 1.
Table 2. Clinical and MRI Results of Study 1:
| Dimethyl Fumarate 240 mg BID | Placebo | P-value | |
|---|---|---|---|
| Clinical Endpoints | N=410 | N=408 | |
| Proportion relapsing (primary endpoint) | 27% | 46% | <0.0001 |
| Relative risk reduction | 49 % | ||
| Annualized relapse rate | 0.172 | 0.364 | <0.0001 |
| Relative reduction | 53 % | ||
| Proportion with disability progression | 16% | 27% | 0.0050 |
| Relative risk reduction | 38 % | ||
| MRI Endpoints | N=152 | N=165 | |
| Mean number of new or newly enlarging T2 lesions over 2 years | 2.6 | 17 | <0.0001 |
| Percentage of subjects with no new or newly enlarging lesions | 45% | 27% | |
| Number of Gd+ lesions at 2 years | 0.1 (0) | 1.8 (0) | |
| Mean (median) Percentage of subjects with | |||
| 0 lesions | 93% | 62% | |
| 1 lesion | 5% | 10% | |
| 2 lesions | <1% | 8% | |
| 3 to 4 lesions | 0 | 9% | |
| 5 or more lesions | <1 % | 11 % | |
| Relative odds reduction (percentage) | 90 % | <0.0001 | |
| Mean number of new T1 hypointense lesions over 2 years | 1.5 | 5.6 | <0.0001 |
Figure 1. Time to 12-Week Confirmed Progression of Disability (Study 1):
Study 2: Placebo-Controlled Trial in RRMS
Study 2 was a 2-year multicenter, randomized, double-blind, placebo-controlled study that also included an open-label comparator arm in patients with RRMS. The primary endpoint was the annualized relapse rate at 2 years. Additional endpoints at 2 years included the number of new or newly enlarging T2 hyperintense lesions, number of T1 hypointense lesions, number of Gd+ lesions, proportion of patients relapsed, and time to confirmed disability progression as defined in Study 1.
Patients were randomized to receive dimethyl fumarate 240 mg twice a day (n=359), dimethyl fumarate 240 mg three times a day (n=345), an open-label comparator (n=350), or placebo (n=363) for up to 2 years. The median age was 37 years, median time since diagnosis was 3 years, and median EDSS score at baseline was 2.5. The median time on study drug for all treatment arms was 96 weeks. The percentages of patients who completed 96 weeks on study drug per treatment group were 70% for patients assigned to dimethyl fumarate 240 mg twice a day, 72% for patients assigned to dimethyl fumarate 240 mg three times a day, and 64% for patients assigned to placebo groups.
Dimethyl fumarate had a statistically significant effect on the relapse and MRI endpoints described above. There was no statistically significant effect on disability progression. The dimethyl fumarate 240 mg three times daily dose resulted in no additional benefit over the dimethyl fumarate 240 mg twice daily dose. The results for this study (240 mg twice a day vs. placebo) are shown in Table 3.
Table 3. Clinical and MRI Results of Study 2:
| Dimethyl Fumarate 240 mg BID | Placebo | P-value | |
|---|---|---|---|
| Clinical Endpoints | N=359 | N=363 | |
| Annualized relapse rate | 0.224 | 0.401 | <0.0001 |
| Relative reduction | 44 % | ||
| Proportion relapsing | 29% | 41 % | 0.0020 |
| Relative risk reduction | 34 % | ||
| Proportion with disability progression | 13% | 17 % | 0.25 |
| Relative risk reduction | 21 % | ||
| MRI Endpoints | N=147 | N=144 | |
| Mean number of new or newly enlarging | 5.1 | 17.4 | <0.0001 |
| T2 lesions over 2 years | |||
| Percentage of subjects with no new or newly enlarging lesions | 27 % | 12 % | |
| Number of Gd+ lesions at 2 years Mean (median) | 0.5 (0.0) | 2.0 (0.0) | |
| Percentage of subjects with | |||
| 0 lesions | 80% | 61% | |
| 1 lesion | 11% | 17% | |
| 2 lesions | 3% | 6% | |
| 3 to 4 lesions | 3% | 2% | |
| 5 or more lesions | 3% | 14 % | |
| Relative odds reduction (percentage) | 74 % | <0.0001 | |
| Mean number of new T1 hypointense lesions over 2 years | 3.0 | 7.0 | <0.0001 |
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.