COMETRIQ Capsule Ref.[27457] Active ingredients: Cabozantinib
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
In vitro biochemical and/or cellular assays have shown that cabozantinib inhibits the tyrosine kinase activity of RET, MET, VEGFR-1, -2 and -3, KIT, TRKB, FLT-3, AXL, ROS1, TYRO3, MER, and TIE-2. These receptor tyrosine kinases are involved in both normal cellular function and pathologic processes such as oncogenesis, metastasis, tumor angiogenesis, drug resistance, and maintenance of the tumor microenvironment.
12.2. Pharmacodynamics
Cardiac Electrophysiology
The effect of orally administered COMETRIQ 140 mg on QTc interval was evaluated in a randomized, double-blinded, placebo-controlled study in patients with MTC. A mean increase in QTcF of 10-15 ms was observed at 4 weeks after initiating COMETRIQ. A concentration-QTc relationship could not be definitively established. Changes in cardiac wave form morphology or new rhythms were not observed. No COMETRIQ-treated patients had a QTcF >500 ms [see Clinical Studies (14)].
12.3. Pharmacokinetics
A population pharmacokinetic analysis of cabozantinib was performed using data collected from 289 patients with solid tumors including MTC following oral administration of 140 mg daily doses. Repeat daily dosing of COMETRIQ at 140 mg for 19 days resulted in 4- to 5-fold mean cabozantinib accumulation (based on AUC) compared to a single dose administration; steady state was achieved by Day 15.
Absorption
Following oral administration of COMETRIQ, median time to peak cabozantinib plasma concentrations (Tmax) ranged from 2 to 5 hours post-dose.
A 19% increase in the Cmax of the tablet formulation (CABOMETYX) compared to the capsule formulation (COMETRIQ) was observed following a single 140 mg dose. A less than 10% difference in the AUC was observed between cabozantinib tablet (CABOMETYX) and capsule (COMETRIQ) formulations [see Dosage and Administration (2.1)].
Cabozantinib Cmax and AUC values increased by 41% and 57%, respectively, following a high-fat meal relative to fasted conditions in healthy subjects administered a single 140 mg oral COMETRIQ dose.
Distribution
The oral volume of distribution (V/F) of cabozantinib is approximately 349 L. Cabozantinib is highly protein bound in human plasma (≥99.7%).
Elimination
The predicted effective half-life is approximately 55 hours and the clearance (CL/F) at steady-state is estimated to be 4.4 L/hr.
Metabolism
Cabozantinib is a substrate of CYP3A4 in vitro.
Excretion
Approximately 81% of the total administered radioactivity was recovered within a 48-day collection period following a single 140 mg dose of an investigational 14C-cabozantinib formulation in healthy subjects. Approximately 54% was recovered in feces and 27% in urine. Unchanged cabozantinib accounted for 43% of the total radioactivity in feces and was not detectable in urine following a 72 hour collection.
Specific Populations
The following patient characteristics did not result in a clinically relevant difference in the pharmacokinetics of cabozantinib: age (20-86 years), sex, race (Whites and non-Whites), or mild to moderate renal impairment (eGFR greater than or equal to 30 mL/min/1.73 m² as estimated by MDRD (modification of diet in renal disease equation)). The pharmacokinetics of cabozantinib is unknown in patients with worse than moderate renal impairment (eGFR less than 29 mL/min/1.73m²) as estimated by MDRD equation or renal impairment requiring dialysis.
Patients with Hepatic Impairment
Following a single oral 60 mg COMETRIQ, mean AUC0-inf for cabozantinib increased by 81% in subjects with mild (Child-Pugh A) hepatic impairment and 63% in subjects with moderate (Child-Pugh B) hepatic impairment compared to subjects with normal hepatic function [see Dosage and Administration (2.5), Use in Specific Populations (8.6)].
The pharmacokinetics of cabozantinib has not been studied in patients with severe (Child-Pugh C) hepatic impairment [see Use in Specific Populations (8.6)].
Drug Interaction Studies
CYP3A4 Inhibition on Cabozantinib
Administration of a strong CYP3A4 inhibitor, ketoconazole (400 mg daily for 27 days) to healthy subjects increased single-dose plasma cabozantinib exposure (AUC0-inf) by 38%.
CYP3A4 Induction on Cabozantinib
Administration of a strong CYP3A4 inducer, rifampin (600 mg daily for 31 days) to healthy subjects decreased single-dose plasma cabozantinib exposure (AUC0-inf) by 77%.
Cabozantinib on CYP2C8 substrates
No clinically-significant effect on single-dose rosiglitazone (a CYP2C8 substrate) plasma exposure (Cmax and AUC) was observed when co-administered with cabozantinib at steady-state plasma concentrations (≥100 mg/day daily for a minimum of 21 days) in patients with solid tumors.
Gastric pH modifying agents on Cabozantinib
No clinically-significant effect on plasma cabozantinib exposure (AUC) was observed following co-administration of the proton pump inhibitor (PPI) esomeprazole (40 mg daily for 6 days) with a single dose of 100 mg cabozantinib to healthy volunteers.
In vitro Studies
Metabolic Pathways
Inhibition of CYP3A4 reduced the formation of the XL184 N-oxide metabolite by >80%. Inhibition of CYP2C9 had a minimal effect on cabozantinib metabolite formation (i.e., a <20% reduction). Inhibition of CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 and CYP2E1 had no effect on cabozantinib metabolite formation.
Although cabozantinib is an inhibitor of CYP2C8 in vitro, a clinical study of this potential interaction concluded that concurrent use did not result in a clinically relevant effect on CYP2C8 substrate exposure. Given this finding, other less sensitive substrates of pathways affected by cabozantinib in vitro (i.e., CYP2C9, CYP2C19, and CYP3A4) were not evaluated in a clinical study because, although a clinically relevant exposure effect cannot be ruled out, it is unlikely. Cabozantinib does not inhibit CYP1A2 and CYP2D6 isozymes in vitro.
Cabozantinib is an inducer of CYP1A1 mRNA; however, the clinical relevance of this finding is unknown. Cabozantinib does not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or CYP3A4.
Drug Transporter Systems
Cabozantinib is an inhibitor, but not a substrate, of P-gp transport activities and has the potential to increase plasma concentrations of co-administered substrates of P-gp. The clinical relevance of this finding is unknown.
Cabozantinib is a substrate of MRP2 in vitro and MRP2 inhibitors have the potential to increase plasma concentrations of cabozantinib. The clinical relevance of this finding is unknown.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of cabozantinib has been evaluated in two species: rasH2 transgenic mice and Sprague-Dawley rats. In the 2-year rat carcinogenicity study, once daily oral administration of cabozantinib resulted in a statistically significant increase in the incidence of malignant/complex malignant pheochromocytoma in combination with benign pheochromocytoma or in benign pheochromocytoma alone in male rats at a dose of 1 mg/kg (approximately 0.6 times the human exposure by AUC at the recommended 140 mg dose). Cabozantinib was not carcinogenic in a 26-week carcinogenicity study in rasH2 transgenic mice at a slightly higher exposure than the intended human therapeutic exposure.
Cabozantinib was not mutagenic in vitro in the bacterial reverse mutation (Ames) assay and was not clastogenic in both the in vitro cytogenetic assay using human lymphocytes or in the in vivo mouse micronucleus assay.
Based on nonclinical findings, male and female fertility may be impaired by treatment with COMETRIQ. In a fertility study in which cabozantinib was administered to male and female rats at doses of 1, 2.5, and 5 mg/kg/day, male fertility was significantly compromised at doses equal to or greater than 2.5 mg/kg/day (approximately equal to the human exposure by AUC at the recommended dose), with a decrease in sperm counts and reproductive organ weights. In females, fertility was significantly reduced at doses equal to or greater than 1 mg/kg/day (approximately 50% of the human exposure by AUC at the 140 mg recommended dose) with a significant decrease in the number of live embryos and a significant increase in pre- and post-implantation losses.
Observations of effects on reproductive tract tissues in general toxicology studies were supportive of effects noted in the dedicated fertility study and included hypospermia and absence of corpora lutea in male and female dogs in a 6-month repeat dose study at exposures equal to 6% and 3%, respectively, the human exposure by AUC at the recommended dose. In addition, female rats administered 5 mg/kg/day for 14 days (approximately equal to the human exposure by AUC at the 140 mg recommended dose) exhibited ovarian necrosis.
14. Clinical Studies
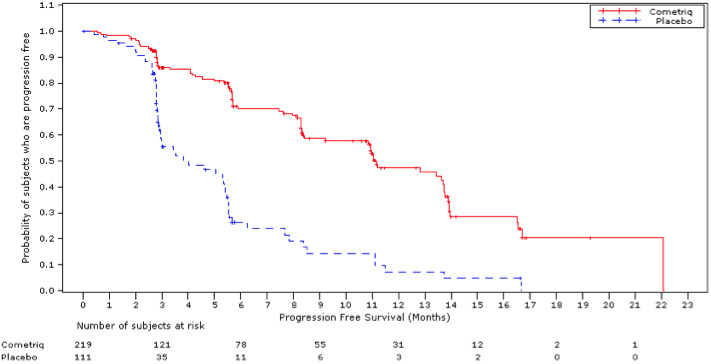
The safety and efficacy of COMETRIQ was assessed in an international, multi-center, randomized, double-blind, controlled trial (Study 1) of 330 patients with metastatic medullary thyroid carcinoma (MTC). Patients were required to have evidence of actively progressive disease within 14 months prior to study entry confirmed by an Independent Radiology Review Committee (IRRC) masked to treatment assignment (89%) or the treating physician (11%). Patients were randomized (2:1) to receive COMETRIQ 140 mg (n = 219) or placebo (n = 111) orally once daily, without food, until disease progression determined by the treating physician or until intolerable toxicity. Randomization was stratified by age (≤ 65 years vs. > 65 years) and prior use of a tyrosine kinase inhibitor (TKI) (yes vs. no). No cross-over was allowed at the time of progression. The main efficacy outcome measures of progression-free survival (PFS), objective response (OR), and response duration were based on IRRC-confirmed events using modified RECIST criteria.
Of 330 patients randomized, 67% were male, the median age was 55 years, 23% were 65 years or older, 89% were white, 54% had a baseline ECOG performance status of 0, and 92% had undergone a thyroidectomy. The RET mutation status determined by a research-use assay was positive in 51%, negative in 14%, and was unknown in 35%. Twenty-five percent (25%) had two or more prior systemic therapies and 21% had been previously treated with a TKI.
A statistically significant prolongation in PFS was demonstrated among COMETRIQ-treated patients compared to those receiving placebo [HR 0.28 (95% CI: 0.19, 0.40); p<0.0001], with median PFS times of 11.2 months and 4.0 months in the COMETRIQ and placebo arms, respectively.
Partial responses were observed only among patients in the COMETRIQ arm (27% vs. 0; p<0.0001). The median duration of objective responses was 14.7 months (95% CI: 11.1, 19.3) for patients treated with COMETRIQ. There was no statistically significant difference in overall survival (median OS: 26.6 months in the COMETRIQ arm vs. 21.1 months in the placebo arm [HR = 0.85 (95% CI: 0.64, 1.12), p = 0.2409]).
Figure 1. Progression-Free Survival:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.