Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: KRKA, d.d., Novo mesto, Šmarješka cesta 6, 8501 Novo mesto, Slovenia
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
Patients with severe (Child-Pugh Class C) hepatic impairment.
Concomitant treatment with any of the following medicinal products given the expected decrease in plasma concentrations of darunavir, ritonavir and cobicistat and the potential for loss of therapeutic effect (see sections 4.4 and 4.5).
Applicable to darunavir boosted with either ritonavir or cobicistat:
Applicable to darunavir boosted with cobicistat, not when boosted with ritonavir:
Darunavir boosted with either ritonavir or cobicistat inhibits the elimination of active substances that are highly dependent on CYP3A for clearance, which results in increased exposure to the co-administered medicinal product. Therefore, concomitant treatment with such medicinal products for which elevated plasma concentrations are associated with serious and/or life-threatening events is contraindicated (applies to darunavir boosted with either ritonavir or cobicistat). These active substances include e.g.:
Regular assessment of virological response is advised. In the setting of lack or loss of virological response, resistance testing should be performed.
Darunavir must always be given orally with cobicistat or low dose ritonavir as a pharmacokinetic enhancer and in combination with other antiretroviral medicinal products (see section 5.2). The Summary of Product Characteristics of cobicistat or ritonavir as appropriate, must therefore be consulted prior to initiation of therapy with darunavir.
Increasing the dose of ritonavir from that recommended in section 4.2 did not significantly affect darunavir concentrations. It is not recommended to alter the dose of cobicistat or ritonavir.
Darunavir binds predominantly to α1-acid glycoprotein. This protein binding is concentration-dependent indicative for saturation of binding. Therefore, protein displacement of medicinal products highly bound to α1-acid glycoprotein cannot be ruled out (see section 4.5).
Darunavir Krka used in combination with cobicistat or low dose ritonavir once daily in ART- experienced patients should not be used in patients with one or more darunavir resistance associated mutations (DRV-RAMs) or HIV-1 RNA ≥100,000 copies/ml or CD4+ cell count <100 cells x 106/l (see section 4.2). Combinations with optimised background regimen (OBRs) other than ≥2 NRTIs have not been studied in this population. Limited data are available in patients with HIV-1 clades other than B (see section 5.1).
Darunavir is not recommended for use in paediatric patients below 3 years of age or less than 15 kg body weight (see sections 4.2 and 5.3).
Darunavir/ritonavir should be used during pregnancy only if the potential benefit justifies the potential risk.
Caution should be used in pregnant women with concomitant medicinal products which may further decrease darunavir exposure (see sections 4.5 and 5.2).
Treatment with darunavir/cobicistat 800/150 mg once daily during the second and third trimester has been shown to result in low darunavir exposure, with a reduction of around 90% in Cmin levels (see section 5.2). Cobicistat levels decrease and may not provide sufficient boosting. The substantial reduction in darunavir exposure may result in virological failure and an increased risk of mother to child transmission of HIV infection. Therefore, therapy with darunavir/cobicistat should not be initiated during pregnancy, and women who become pregnant during therapy with darunavir/cobicistat should be switched to an alternative regimen (see sections 4.2 and 4.6). Darunavir given with low dose ritonavir may be considered as an alternative.
As limited information is available on the use of darunavir in patients aged 65 and over, caution should be exercised in the administration of darunavir in elderly patients, reflecting the greater frequency of decreased hepatic function and of concomitant disease or other therapy (see sections 4.2 and 5.2).
During the darunavir/ritonavir clinical development program (N=3,063), severe skin reactions, which may be accompanied with fever and/or elevations of transaminases, have been reported in 0.4% of patients. DRESS (Drug Rash with Eosinophilia and Systemic Symptoms) and Stevens-Johnson Syndrome has been rarely (<0.1%) reported, and during post-marketing experience toxic epidermal necrolysis and acute generalised exanthematous pustulosis have been reported. Darunavir should be discontinued immediately if signs or symptoms of severe skin reactions develop. These can include, but are not limited to, severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, blisters, oral lesions, conjunctivitis, hepatitis and/or eosinophilia.
Rash occurred more commonly in treatment-experienced patients receiving regimens containing darunavir/ritonavir + raltegravir compared to patients receiving darunavir/ritonavir without raltegravir or raltegravir without darunavir (see section 4.8).
Darunavir contains a sulphonamide moiety. Darunavir should be used with caution in patients with a known sulphonamide allergy.
Drug-induced hepatitis (e.g. acute hepatitis, cytolytic hepatitis) has been reported with darunavir. During the darunavir/ritonavir clinical development program (N=3,063), hepatitis was reported in 0.5% of patients receiving combination antiretroviral therapy with darunavir/ritonavir. Patients with pre-existing liver dysfunction, including chronic active hepatitis B or C, have an increased risk for liver function abnormalities including severe and potentially fatal hepatic adverse reactions. In case of concomitant antiviral therapy for hepatitis B or C, please refer to the relevant product information for these medicinal products.
Appropriate laboratory testing should be conducted prior to initiating therapy with darunavir used in combination with cobicistat or low dose ritonavir and patients should be monitored during treatment. Increased aspartate aminotransferase/alanine aminotransferase (AST/ALT) monitoring should be considered in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases, especially during the first several months of darunavir used in combination with cobicistat or low dose ritonavir treatment.
If there is evidence of new or worsening liver dysfunction (including clinically significant elevation of liver enzymes and/or symptoms such as fatigue, anorexia, nausea, jaundice, dark urine, liver tenderness, hepatomegaly) in patients using darunavir used in combination with cobicistat or low dose ritonavir, interruption or discontinuation of treatment should be considered promptly.
The safety and efficacy of darunavir have not been established in patients with severe underlying liver disorders and darunavir is therefore contraindicated in patients with severe hepatic impairment. Due to an increase in the unbound darunavir plasma concentrations, darunavir should be used with caution in patients with mild or moderate hepatic impairment (see sections 4.2, 4.3 and 5.2).
No special precautions or dose adjustments for darunavir/ritonavir are required in patients with renal impairment. As darunavir and ritonavir are highly bound to plasma proteins, it is unlikely that they will be significantly removed by haemodialysis or peritoneal dialysis. Therefore, no special precautions or dose adjustments are required in these patients (see sections 4.2 and 5.2). Cobicistat has not been studied in patients receiving dialysis, therefore, no recommendation can be made for the use of darunavir/cobicistat in these patients (see section 4.2).
Cobicistat decreases the estimated creatinine clearance due to inhibition of tubular secretion of creatinine. This should be taken into consideration if darunavir with cobicistat is administered to patients in whom the estimated creatinine clearance is used to adjust doses of co-administered medicinal products (see section 4.2 and cobicistat SmPC).
There are currently inadequate data to determine whether co-administration of tenofovir disoproxil and cobicistat is associated with a greater risk of renal adverse reactions compared with regimens that include tenofovir disoproxil without cobicistat.
There have been reports of increased bleeding, including spontaneous skin haematomas and haemarthrosis in patients with haemophilia type A and B treated with PIs. In some patients additional factor VIII was given. In more than half of the reported cases, treatment with PIs was continued or reintroduced if treatment had been discontinued. A causal relationship has been suggested, although the mechanism of action has not been elucidated. Haemophiliac patients should, therefore, be made aware of the possibility of increased bleeding.
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
In HIV infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections and pneumonia caused by Pneumocystis jirovecii (formerly known as Pneumocystis carinii). Any inflammatory symptoms should be evaluated and treatment instituted when necessary. In addition, reactivation of herpes simplex and herpes zoster has been observed in clinical studies with darunavir co-administered with low dose ritonavir.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.8).
Several of the interaction studies have been performed with darunavir at lower than recommended doses. The effects on co-administered medicinal products may thus be underestimated and clinical monitoring of safety may be indicated. For full information on interactions with other medicinal products see section 4.5.
Darunavir has different interaction profiles depending on whether the compound is boosted with ritonavir or cobicistat:
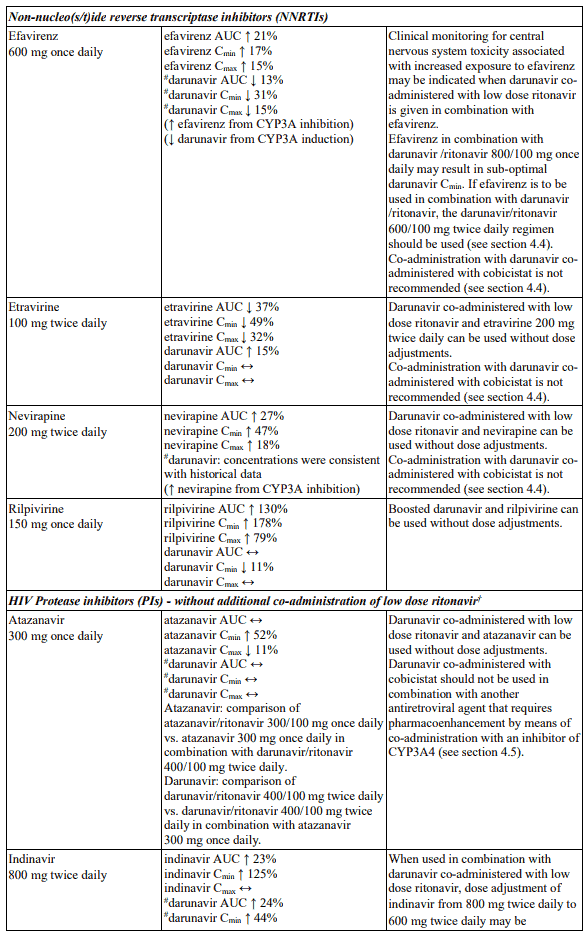
Efavirenz in combination with darunavir may result in sub-optimal darunavir Cmin. If efavirenz is to be used in combination with darunavir, the darunavir/ritonavir 600/100 mg twice daily regimen should be used. See the Summary of Product Characteristics for Darunavir Krka 600 mg tablets (see section 4.5).
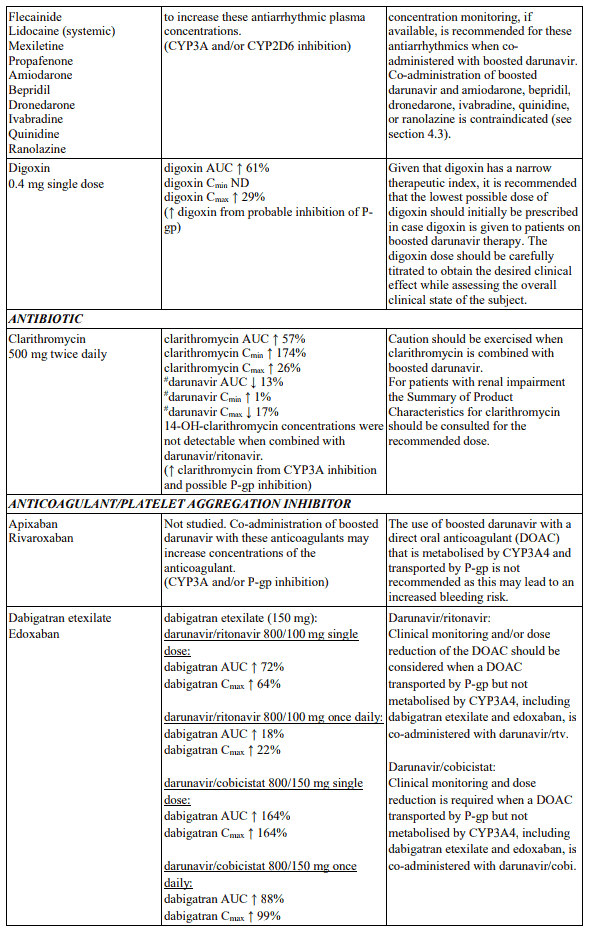
Life-threatening and fatal drug interactions have been reported in patients treated with colchicine and strong inhibitors of CYP3A and P-glycoprotein (P-gp; see sections 4.3 and 4.5).
The interaction profile of darunavir may differ depending on whether ritonavir or cobicistat is used as pharmacoenhancer. The recommendations given for concomitant use of darunavir and other medicinal products may therefore differ depending on whether darunavir is boosted with ritonavir or cobicistat (see sections 4.3 and 4.4), and caution is also required during the first time of treatment if switching the pharmacoenhancer from ritonavir to cobicistat (see section 4.4).
Darunavir and ritonavir are metabolised by CYP3A. Medicinal products that induce CYP3A activity would be expected to increase the clearance of darunavir and ritonavir, resulting in lowered plasma concentrations of these compounds and consequently that of darunavir, leading to loss of therapeutic effect and possible development of resistance (see sections 4.3 and 4.4). CYP3A inducers that are contraindicated include rifampicin, St John’s wort and lopinavir.
Co-administration of darunavir and ritonavir with other medicinal products that inhibit CYP3A may decrease the clearance of darunavir and ritonavir, which may result in increased plasma concentrations of darunavir and ritonavir. Co-administration with strong CYP3A4 inhibitors is not recommended and caution is warranted, these interactions are described in the interaction table below (e.g. indinavir, azole antifungals like clotrimazole).
Darunavir and cobicistat are metabolised by CYP3A, and co-administration with CYP3A inducers may therefore result in subtherapeutic plasma exposure to darunavir. Darunavir boosted with cobicistat is more sensitive to CYP3A induction than ritonavir-boosted darunavir: co-administration of darunavir/cobicistat with medicinal products that are strong inducers of CYP3A (e.g. St John’s wort, rifampicin, carbamazepine, phenobarbital, and phenytoin) is contraindicated (see section 4.3). Co-administration of darunavir/cobicistat with weak to moderate CYP3A inducers (e.g. efavirenz, etravirine, nevirapine, boceprevir, fluticasone, and bosentan) is not recommended (see interaction table below).
For co-administration with strong CYP3A4 inhibitors, the same recommendations apply independent of whether darunavir is boosted with ritonavir or with cobicistat (see section above).
Darunavir and ritonavir are inhibitors of CYP3A, CYP2D6 and P-gp. Co-administration of darunavir/ritonavir with medicinal products primarily metabolised by CYP3A and/or CYP2D6 or transported by P-gp may result in increased systemic exposure to such medicinal products, which could increase or prolong their therapeutic effect and adverse reactions.
Darunavir co-administered with low dose ritonavir must not be combined with medicinal products that are highly dependent on CYP3A for clearance and for which increased systemic exposure is associated with serious and/or life-threatening events (narrow therapeutic index) (see section 4.3).
Co-administration of boosted darunavir with drugs that have active metabolite(s) formed by CYP3A may result in reduced plasma concentrations of these active metabolite(s), potentially leading to loss of their therapeutic effect (see the Interaction table below).
The overall pharmacokinetic enhancement effect by ritonavir was an approximate 14-fold increase in the systemic exposure of darunavir when a single dose of 600 mg darunavir was given orally in combination with ritonavir at 100 mg twice daily. Therefore, darunavir must only be used in combination with a pharmacokinetic enhancer (see sections 4.4 and 5.2).
A clinical study utilising a cocktail of medicinal products that are metabolised by cytochromes CYP2C9, CYP2C19 and CYP2D6 demonstrated an increase in CYP2C9 and CYP2C19 activity and inhibition of CYP2D6 activity in the presence of darunavir/ritonavir, which may be attributed to the presence of low dose ritonavir. Co-administration of darunavir and ritonavir with medicinal products which are primarily metabolised by CYP2D6 (such as flecainide, propafenone, metoprolol) may result in increased plasma concentrations of these medicinal products, which could increase or prolong their therapeutic effect and adverse reactions. Co-administration of darunavir and ritonavir and medicinal products primarily metabolised by CYP2C9 (such as warfarin) and CYP2C19 (such as methadone) may result in decreased systemic exposure to such medicinal products, which could decrease or shorten their therapeutic effect.
Although the effect on CYP2C8 has only been studied in vitro, co-administration of darunavir and ritonavir and medicinal products primarily metabolised by CYP2C8 (such as paclitaxel, rosiglitazone, repaglinide) may result in decreased systemic exposure to such medicinal products, which could decrease or shorten their therapeutic effect.
Ritonavir inhibits the transporters P-glycoprotein, OATP1B1 and OATP1B3, and co-administration with substrates of these transporters can result in increased plasma concentrations of these compounds (e.g. dabigatran etexilate, digoxin, statins and bosentan; see the Interaction table below).
The recommendations for darunavir boosted with ritonavir are adequate also for darunavir boosted with cobicistat with regard to substrates of CYP3A4, CYP2D6, P-glycoprotein, OATP1B1 and OATP1B3 (see contraindications and recommendations presented in the section above). Cobicistat 150 mg given with darunavir 800 mg once daily enhances darunavir pharmacokinetic parameters in a comparable way to ritonavir (see section 5.2).
Unlike ritonavir, cobicistat does not induce CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 or UGT1A1. For further information on cobicistat, consult the cobicistat Summary of Product Characteristics.
Interaction studies have only been performed in adults.
Several of the interaction studies (indicated by # in the table below) have been performed at lower than recommended doses of darunavir or with a different dosing regimen (see section 4.2 Posology). The effects on co-administered medicinal products may thus be underestimated and clinical monitoring o safety may be indicated.
The interaction profile of darunavir depends on whether ritonavir or cobicistat is used as pharmacokinetic enhancer. Darunavir may therefore have different recommendations for concomitant medications depending on whether the compound is boosted with ritonavir or cobicistat. No interaction studies presented in the table have been performed with darunavir boosted with cobicistat. The same recommendations apply, unless specifically indicated. For further information on cobicistat, consult the cobicistat Summary of Product Characteristics.
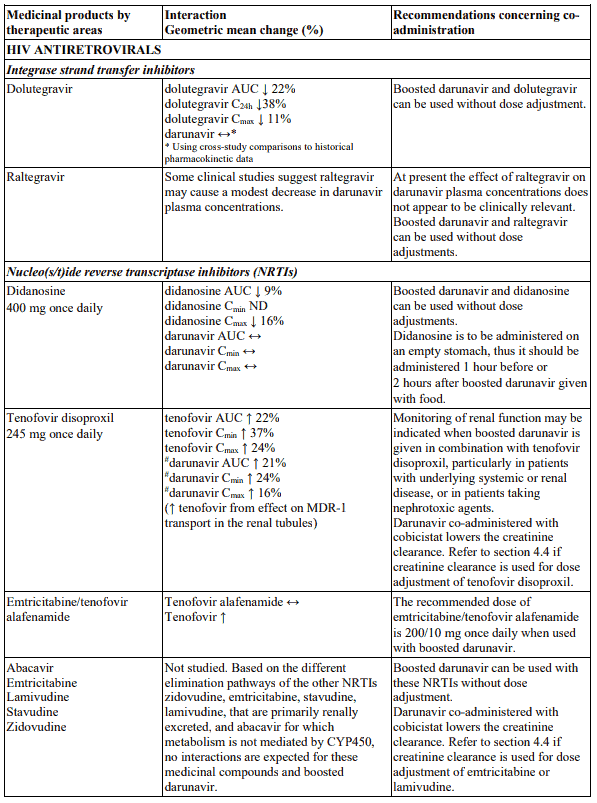
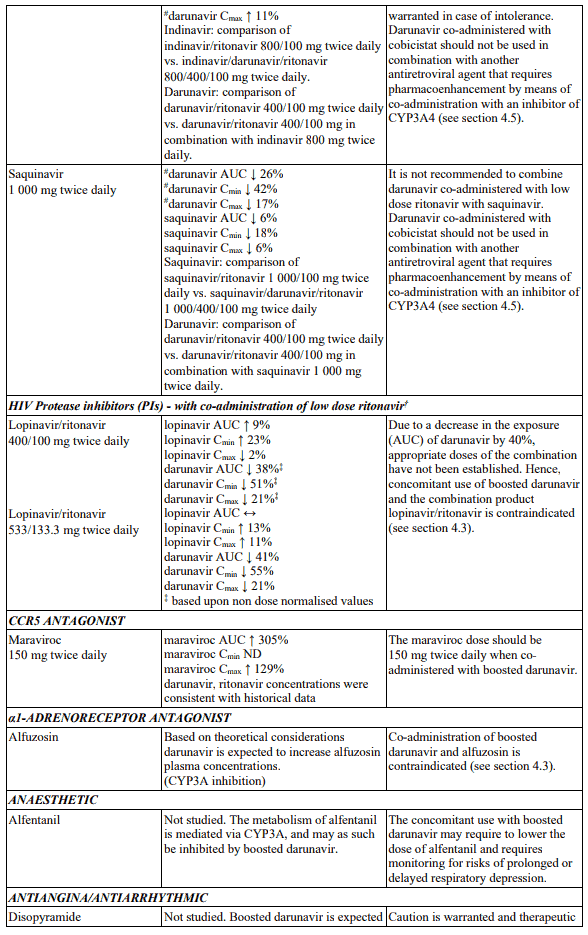
Interactions between darunavir/ritonavir and antiretroviral and non-antiretroviral medicinal products are listed in the table below. The direction of the arrow for each pharmacokinetic parameter is based on the 90% confidence interval of the geometric mean ratio being within (↔), below (↓) or above (↑) the 80-125% range (not determined as “ND”).
In the table below the specific pharmacokinetic enhancer is specified when recommendations differ. When the recommendation is the same for darunavir when co-administered with a low dose ritonavir or cobicistat, the term “boosted darunavir” is used.
The below list of examples of drug-drug interactions is not comprehensive and therefore the label of each drug that is co-administered with darunavir should be consulted for information related to the route of metabolism, interaction pathways, potential risks, and specific actions to be taken with regards to co-administration.
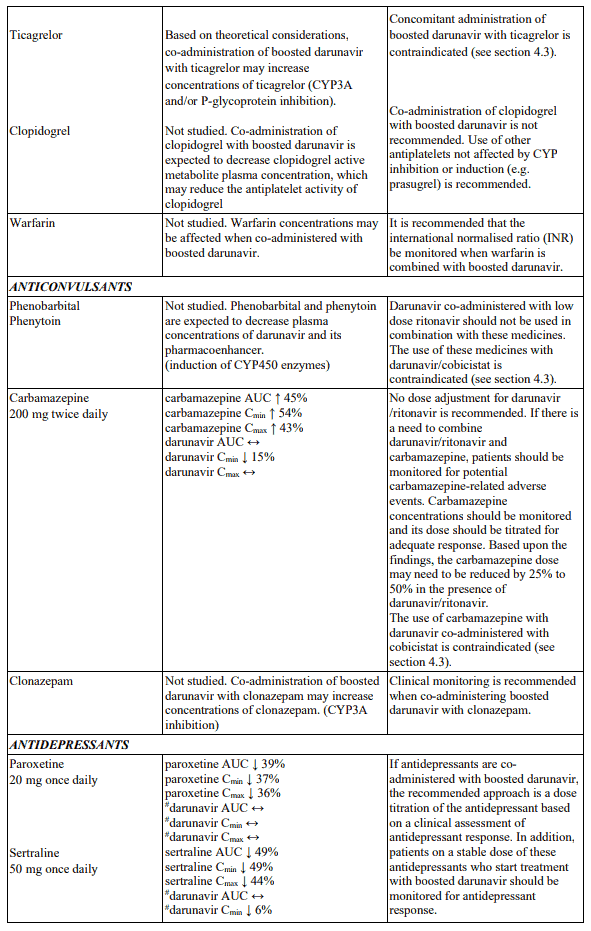
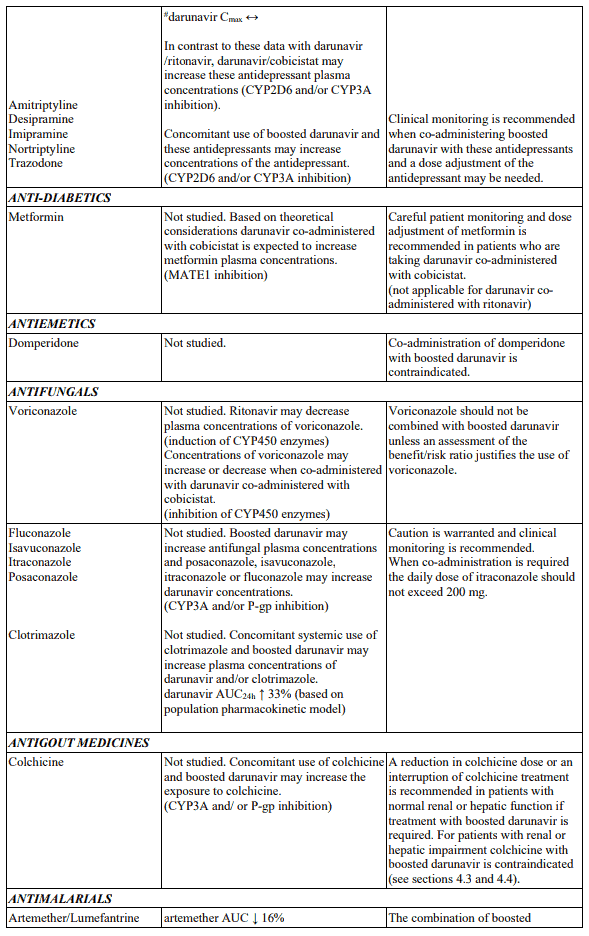
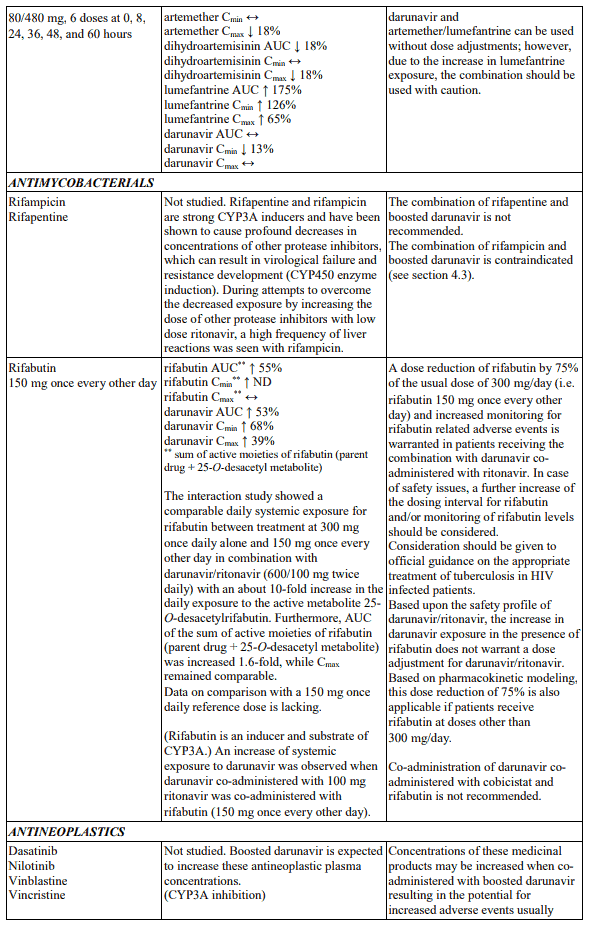
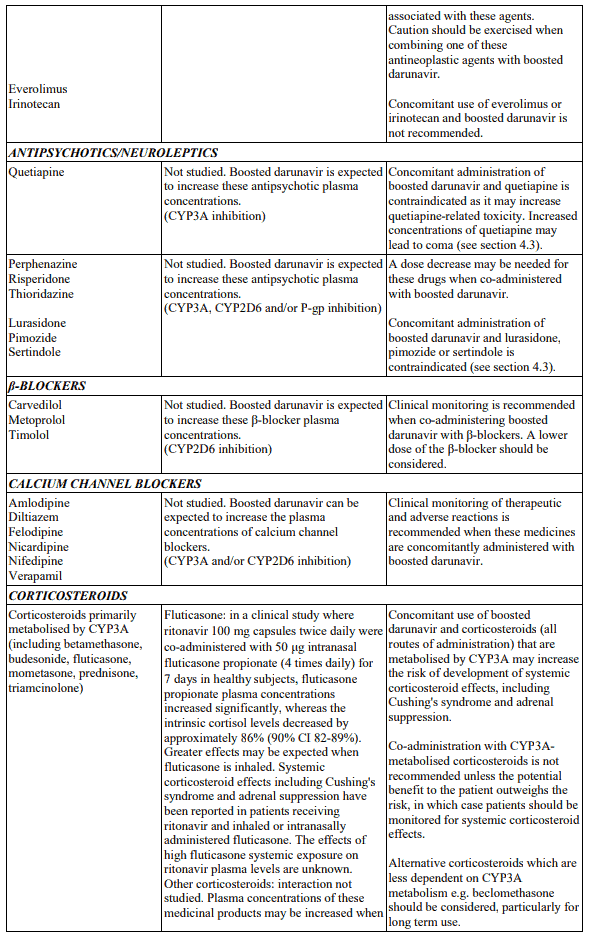
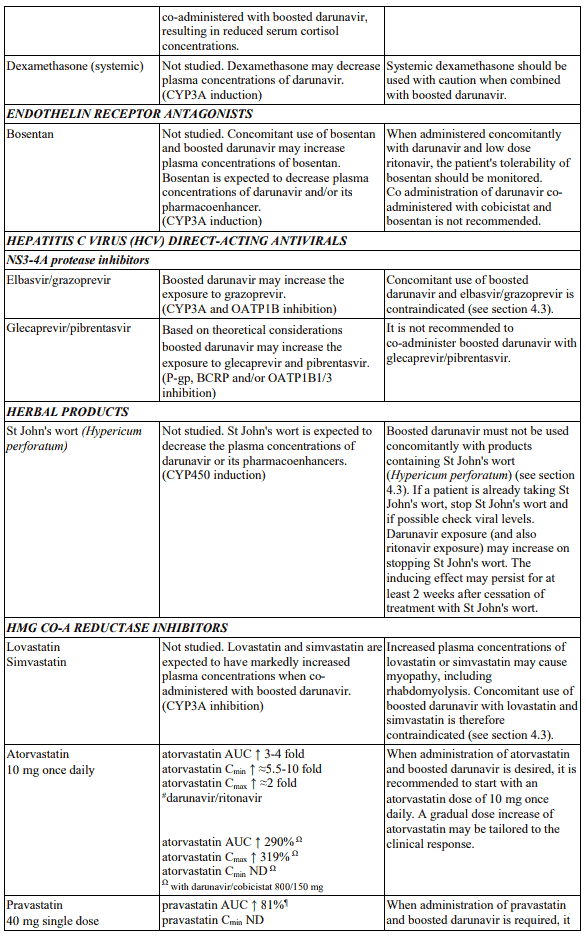
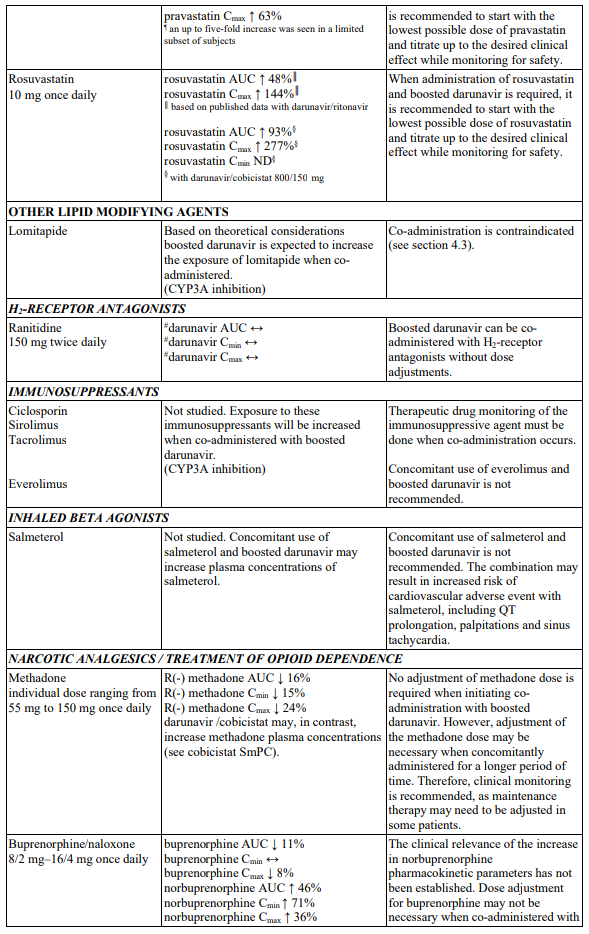
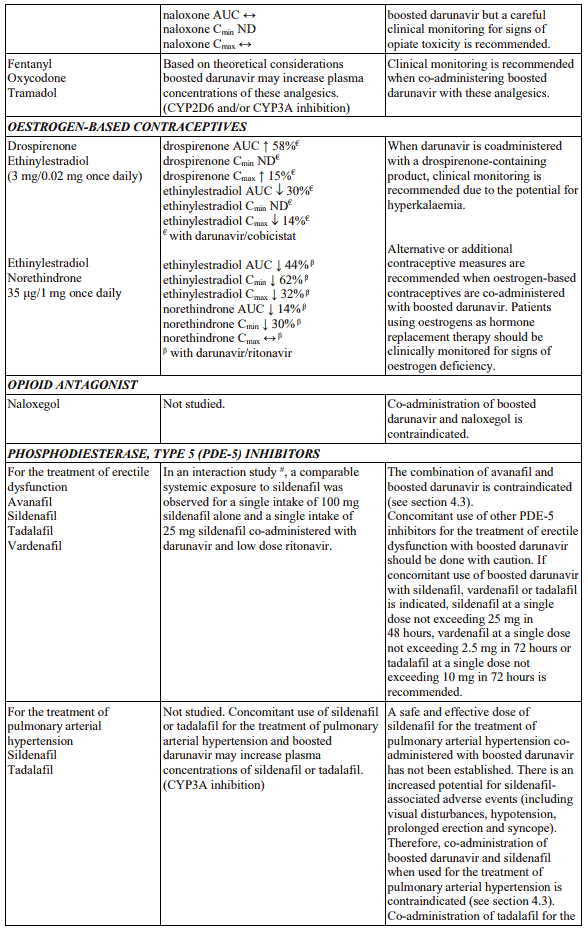
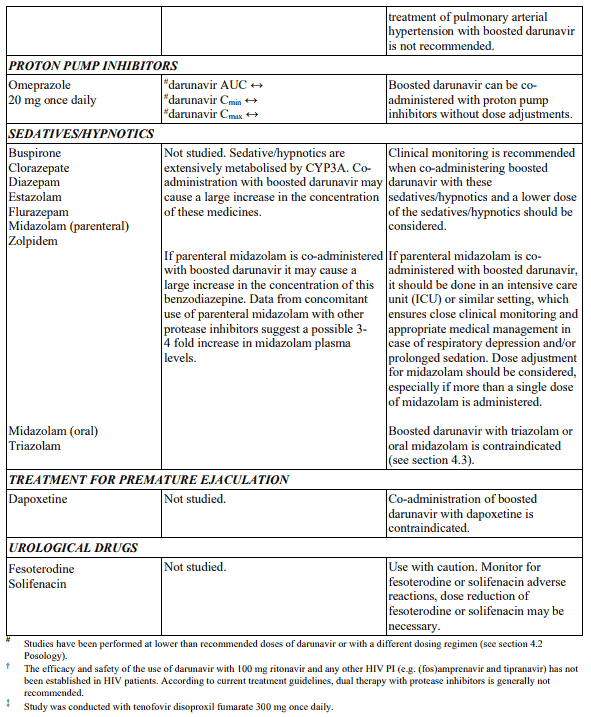
Table 1. Interactions and dose recommendations with medicinal products:
As a general rule, when deciding to use antiretroviral agents for the treatment of HIV infection in pregnant women and consequently for reducing the risk of HIV vertical transmission to the newborn, the animal data as well as the clinical experience in pregnant women should be taken into account.
There are no adequate and well controlled studies on pregnancy outcome with darunavir in pregnant women. Studies in animals do not indicate direct harmful effects with respect to pregnancy, embryonal/foetal development, parturition or postnatal development (see section 5.3).
Darunavir co-administered low dose ritonavir should be used during pregnancy only if the potential benefit justifies the potential risk.
Treatment with darunavir/cobicistat 800/150 mg during pregnancy results in low darunavir exposure (see section 5.2), which may be associated with an increased risk of treatment failure and an increased risk of HIV transmission to the child. Therapy with darunavir/cobicistat should not be initiated during pregnancy, and women who become pregnant during therapy with darunavir/cobicistat should be switched to an alternative regimen (see sections 4.2 and 4.4).
It is not known whether darunavir is excreted in human milk. Studies in rats have demonstrated that darunavir is excreted in milk and at high levels (1 000 mg/kg/day) resulted in toxicity of the offspring.
Because of the potential for adverse reactions in breast-fed infants, women should be instructed not to breast-feed if they are receiving Darunavir Krka.
In order to avoid transmission of HIV to the infant it is recommended that women living with HIV do not breast-feed.
No human data on the effect of darunavir on fertility are available. There was no effect on mating or fertility with darunavir treatment in rats (see section 5.3).
Darunavir in combination with cobicistat or ritonavir has no or negligible influence on the ability to drive and use machines. However, dizziness has been reported in some patients during treatment with regimens containing darunavir co-administered with cobicistat or low dose ritonavir and should be borne in mind when considering a patient’s ability to drive or operate machinery (see section 4.8).
During the clinical development program (N=2 613 treatment-experienced subjects who initiated therapy with darunavir/ritonavir 600/100 mg twice daily), 51.3% of subjects experienced at least one adverse reaction. The total mean treatment duration for subjects was 95.3 weeks. The most frequent adverse reactions reported in clinical studies and as spontaneous reports are diarrhoea, nausea, rash, headache and vomiting. The most frequent serious reactions are acute renal failure, myocardial infarction, immune reconstitution inflammatory syndrome, thrombocytopenia, osteonecrosis, diarrhoea, hepatitis and pyrexia.
In the 96 week analysis, the safety profile of darunavir/ritonavir 800/100 mg once daily in treatmentnaïve subjects was similar to that seen with darunavir/ritonavir 600/100 mg twice daily in treatmentexperienced subjects except for nausea which was observed more frequently in treatment-naïve subjects. This was driven by mild intensity nausea. No new safety findings were identified in the 192 week analysis of the treatment-naïve subjects in which the mean treatment duration of darunavir/ritonavir 800/100 mg once daily was 162.5 weeks.
During the Phase III clinical studies GS-US-216-130 with darunavir/cobicistat (N=313 treatment-naïve and treatment-experienced subjects), 66.5% of subjects experienced at least one adverse reaction. The mean treatment duration was 58.4 weeks. The most frequent adverse reactions reported were diarrhoea (28%), nausea (23%), and rash (16%). Serious adverse reactions are diabetes mellitus, (drug) hypersensitivity, immune reconstitution inflammatory syndrome, rash and vomiting.
For information on cobicistat, consult the cobicistat Summary of Product Characteristics.
Adverse reactions are listed by system organ class (SOC) and frequency category. Within each frequency category, adverse reactions are presented in order of decreasing seriousness. Frequency categories are defined as follows: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1 000 to <1/100), rare (≥1/10 000 to <1/1 000) and not known (frequency cannot be estimated from the available data).
Table 2. Adverse reactions observed with darunavir/ritonavir in clinical studies and post-marketing:
| MedDRA system organ class Frequency category | Adverse reaction |
|---|---|
| Infections and infestations | |
| uncommon | herpes simplex |
| Blood and lymphatic system disorders | |
| uncommon | thrombocytopenia, neutropenia, anaemia, leukopenia |
| rare | increased eosinophil count |
| Immune system disorders | |
| uncommon | immune reconstitution inflammatory syndrome, (drug) hypersensitivity |
| Endocrine disorders | |
| uncommon | hypothyroidism, increased blood thyroid stimulating hormone |
| Metabolism and nutrition disorders | |
| common | diabetes mellitus, hypertriglyceridaemia, hypercholesterolaemia, hyperlipidaemia |
| uncommon | gout, anorexia, decreased appetite, decreased weight, increased weight, hyperglycaemia, insulin resistance, decreased high density lipoprotein, increased appetite, polydipsia, increased blood lactate dehydrogenase |
| Psychiatric disorders | |
| common | insomnia |
| uncommon | depression, disorientation, anxiety, sleep disorder, abnormal dreams, nightmare, decreased libido |
| rare | confusional state, altered mood, restlessness |
| Nervous system disorders | |
| common | headache, peripheral neuropathy, dizziness |
| uncommon | lethargy, paraesthesia, hypoaesthesia, dysgeusia, disturbance in attention, memory impairment, somnolence |
| rare | syncope, convulsion, ageusia, sleep phase rhythm disturbance |
| Eye disorders | |
| uncommon | conjunctival hyperaemia, dry eye |
| rare | visual disturbance |
| Ear and labyrinth disorders | |
| uncommon | vertigo |
| Cardiac disorders | |
| uncommon | myocardial infarction, angina pectoris, prolonged electrocardiogram QT, tachycardia rare acute myocardial infarction, sinus bradycardia, palpitations |
| Vascular disorders | |
| uncommon | hypertension, flushing |
| Respiratory, thoracic and mediastinal disorders | |
| uncommon | dyspnoea, cough, epistaxis, throat irritation |
| rare | rhinorrhoea |
| Gastrointestinal disorders | |
| very common | diarrhoea |
| common | vomiting, nausea, abdominal pain, increased blood amylase, dyspepsia, abdominal distension, flatulence |
| uncommon | pancreatitis, gastritis, gastrooesophageal reflux disease, aphthous stomatitis, retching, dry mouth, abdominal discomfort, constipation, increased lipase, eructation, oral dysaesthesia |
| rare | stomatitis, haematemesis, cheilitis, dry lip, coated tongue |
| Hepatobiliary disorders | |
| common | increased alanine aminotransferase |
| uncommon | hepatitis, cytolytic hepatitis, hepatic steatosis, hepatomegaly, increased transaminase, increased aspartate aminotransferase, increased blood bilirubin, increased blood alkaline phosphatase, increased gamma-glutamyltransferase |
| Skin and subcutaneous tissue disorders | |
| common | rash (including macular, maculopapular, papular, erythematous and pruritic rash), pruritus |
| uncommon | angioedema, generalised rash, allergic dermatitis, urticaria, eczema, erythema, hyperhidrosis, night sweats, alopecia, acne, dry skin, nail pigmentation |
| rare | DRESS, Stevens-Johnson syndrome, erythema multiforme, dermatitis, seborrhoeic dermatitis, skin lesion, xeroderma |
| not known | toxic epidermal necrolysis, acute generalised exanthematous pustulosis |
| Musculoskeletal and connective tissue disorders | |
| uncommon | myalgia, osteonecrosis, muscle spasms, muscular weakness, arthralgia, pain in extremity, osteoporosis, increased blood creatine phosphokinase |
| rare | musculoskeletal stiffness, arthritis, joint stiffness |
| Renal and urinary disorders | |
| uncommon | acute renal failure, renal failure, nephrolithiasis, increased blood creatinine, proteinuria, bilirubinuria, dysuria, nocturia, pollakiuria rare decreased creatinine renal clearance |
| rare | crystal nephropathy§ |
| Reproductive system and breast disorders | |
| uncommon | erectile dysfunction, gynaecomastia |
| General disorders and administration site conditions | |
| common | asthenia, fatigue |
| uncommon | pyrexia, chest pain, peripheral oedema, malaise, feeling hot, irritability, pain |
| rare | chills, abnormal feeling, xerosis |
§ adverse reaction identified in the post-marketing setting. Per the guideline on Summary of Product Characteristics (Revision 2, September 2009), the frequency of this adverse reaction in the post-marketing setting was determined using the “Rule of 3”.
Table 3. Adverse reactions observed with darunavir/cobicistat in adult patients:
| MedDRA system organ class Frequency category | Adverse reaction |
|---|---|
| Immune system disorders | |
| common (drug) | hypersensitivity |
| uncommon | immune reconstitution inflammatory syndrome |
| Metabolism and nutrition disorders | |
| common | anorexia, diabetes mellitus, hypercholesterolaemia, hypertriglyceridaemia, hyperlipidaemia |
| Psychiatric disorders | |
| common | abnormal dreams |
| Nervous system disorders | |
| very common | headache |
| Gastrointestinal disorders | |
| very common | diarrhoea, nausea |
| common | vomiting, abdominal pain, abdominal distension, dyspepsia, flatulence, pancreatic enzymes increased |
| uncommon | pancreatitis acute |
| Hepatobiliary disorders | |
| common | hepatic enzyme increased |
| uncommon | hepatitis*, cytolytic hepatitis* |
| Skin and subcutaneous tissue disorders | |
| very common | rash (including macular, maculopapular, papular, erythematous, pruritic rash, generalised rash, and allergic dermatitis) |
| common | angioedema, pruritus, urticaria |
| rare | drug reaction with eosinophilia and systemic symptoms*, Stevens-Johnson syndrome* |
| not known | toxic epidermal necrolysis*, acute generalised exanthematous pustulosis* |
| Musculoskeletal and connective tissue disorders | |
| common | myalgia |
| uncommon | osteonecrosis* |
| Renal and urinary disorders | |
| rare | crystal nephropathy§* |
| Reproductive system and breast disorders | |
| uncommon | gynaecomastia* |
| General disorders and administration site conditions | |
| common | fatigue |
| uncommon | asthenia |
| Investigations | |
| common | increased blood creatinine |
* these adverse drug reactions have not been reported in clinical study experience with darunavir/cobicistat but have been noted with darunavir/ritonavir treatment and could be expected with darunavir/cobicistat too.
§ adverse reaction identified in the post-marketing setting. Per the guideline on Summary of Product Characteristics (Revision 2, September 2009), the frequency of this adverse reaction in the post-marketing setting was determined using the “Rule of 3”.
In clinical studies, rash was mostly mild to moderate, often occurring within the first four weeks of treatment and resolving with continued dosing. In cases of severe skin reaction see the warning in section 4.4. In a single arm study investigating darunavir 800 mg once daily in combination with cobicistat 150 mg once daily and other antiretrovirals 2.2% of patients discontinued treatment due to rash.
During the clinical development program of raltegravir in treatment-experienced patients, rash, irrespective of causality, was more commonly observed with regimens containing darunavir/ritonavir + raltegravir compared to those containing darunavir/ritonavir without raltegravir or raltegravir without darunavir/ritonavir. Rash considered by the investigator to be drug-related occurred at similar rates. The exposure-adjusted rates of rash (all causality) were 10.9, 4.2, and 3.8 per 100 patient-years (PYR), respectively; and for drug-related rash were 2.4, 1.1, and 2.3 per 100 PYR, respectively. The rashes observed in clinical studies were mild to moderate in severity and did not result in discontinuation of therapy (see section 4.4).
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
Increased CPK, myalgia, myositis and rarely, rhabdomyolysis have been reported with the use of protease inhibitors, particularly in combination with NRTIs.
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
In HIV infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
There have been reports of increased spontaneous bleeding in haemophiliac patients receiving antiretroviral protease inhibitors (see section 4.4).
The safety assessment of darunavir with ritonavir in paediatric patients is based on the 48-week analysis of safety data from three Phase II studies. The following patient populations were evaluated (see section 5.1):
Overall, the safety profile in these paediatric patients was similar to that observed in the adult population.
Among 1 968 treatment-experienced patients receiving darunavir co-administered with ritonavir 600/100 mg twice daily, 236 patients were co-infected with hepatitis B or C. Co-infected patients were more likely to have baseline and treatment emergent hepatic transaminase elevations than those without chronic viral hepatitis (see section 4.4).
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.