DEPAKENE Oral solution Ref.[50409] Active ingredients: Valproic acid
Source: Health Products and Food Branch (CA) Revision Year: 2022
Contraindications
DEPAKENE (valproic acid) is contraindicated in:
- Patients with known hypersensitivity to the drug, any ingredient in the formulation or component of the container. For a complete listing of ingredients (see 6 DOSAGE FORMS, STRENGTHS, COMPOSITION AND PACKAGING).
- The treatment of epilepsy
- in pregnancy unless there is no suitable alternative treatment (see 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program, and 7.1.1 Pregnant Women).
- in women of childbearing potential, unless the conditions of the Pregnancy Prevention Program are fulfilled (see 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program, and 7.1.1 Pregnant Women).
- Patients with hepatic disease or significant hepatic dysfunction (see 3 SERIOUS WARNINGS AND PRECAUTIONS BOX, Hepatotoxicity and 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Serious or Fatal Hepatotoxicity).
- Patients known to have mitochondrial disorders caused by mutations in mitochondrial DNA polymerase γ (POLG; e.g., Alpers-Huttenlocher Syndrome) and children under two years of age who are suspected of having a POLG-related disorder (see 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic).
- Patients with known urea cycle disorders (see 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism, Urea Cycle Disorders).
- Patients with known porphyria.
Warnings and precautions
Pregnancy Prevention Program
Valproate has a high teratogenic potential and children exposed in utero to valproate have a high risk for major congenital malformations and neurodevelopmental disorders (see 7.1.1 Pregnant Women). DEPAKENE is contraindicated in the treatment of epilepsy during pregnancy unless no other suitable alternative can be found (see 2 CONTRAINDICATIONS).
Conditions of Pregnancy Prevention Program
The prescriber must ensure that:
- individual circumstances are evaluated in each case and discussed with the patient. This is to guarantee the patient's engagement and understanding of the therapeutic options together with the risks and the measures needed to mitigate the risks.
- the potential for pregnancy is assessed for all female patients.
- the patient understands and acknowledges the risks of major congenital malformations and neurodevelopmental disorders including the magnitude of these risks for children exposed to valproate in utero.
- the patient understands the need to undergo pregnancy testing prior to initiation of treatment and during treatment with valproate as deemed necessary by the patient or treating physician. It is recommended that testing be done following a missed period, the failure of the selected method of contraception or as needed.
- the patient is counselled regarding contraception, and that the patient is capable of complying with the need to use effective and reliable contraception (see 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program, Contraception), without interruption during the entire duration of treatment with valproate.
- the patient understands the need for regular (at least annual) review of treatment by a specialist experienced in the management of epilepsy.
- the patient understands the need to consult her physician as soon as she is planning pregnancy to ensure timely discussion and switching to alternative treatment options prior to conception, and before contraception is discontinued.
- the patient understands the need to urgently consult her physician if she becomes pregnant.
- the patient has received the patient guide.
- the patient has acknowledged that she has understood the risks associated with valproate use and necessary precautions to be taken during treatment (Annual Risk Acknowledgement Form).
These conditions also apply to women who are not currently sexually active unless the prescriber considers that there are compelling and convincing reasons to indicate that there is no risk of pregnancy.
Pharmacist or other health care professional must ensure that:
- the patient card is provided with every valproate dispensing and that the patients understand its content.
- the patient is advised not to stop valproate treatment and to immediately contact a specialist in case of planned or suspected pregnancy.
Female Children
- The prescriber must ensure that parents/caregivers of female children understand the need to contact the specialist once the female child receiving valproate treatment experiences menarche.
- The prescriber must ensure that parents/caregivers of female children who have experienced menarche are provided with comprehensive information about the risks of major congenital malformations and neurodevelopmental disorders including the magnitude of these risks for children exposed to valproate in utero.
- In patients who have experienced menarche, the prescribing specialist must reassess the need for continuing valproate therapy annually and consider alternative treatment options. If valproate is the only suitable treatment, the patient must use at least one effective and reliable method of contraception (preferably a user-independent form) or two complementary forms of contraception. Patient must also meet all other conditions of the Pregnancy Prevention Program. Every effort should be made by the specialist to switch the female children to alternative treatment before they reach adulthood.
Pregnancy Test
Pregnancy must be excluded before the start of treatment with valproate. Treatment must not be initiated in women of child bearing potential without a negative plasma pregnancy test result, confirmed by a health care provider, to rule out unintended use in pregnancy.
Contraception
Women of childbearing potential who are prescribed valproate must use at least one form of effective and reliable contraception (preferably a user-independent form) or two complementary forms of contraception without interruption during the entire duration of treatment. These patients must be provided with comprehensive information on pregnancy prevention and should be referred for contraceptive advice if they are not using effective contraception. Individual circumstances should be evaluated in each case, when choosing the contraception method involving the patient in the discussion, to guarantee her engagement and compliance with the chosen measures. Even if she has amenorrhea she must follow all the advice on effective and reliable contraception. At least 1 of these forms of contraception must be a primary form, which include tubal ligation, partner's vasectomy, intrauterine devices, birth control pills, and topical/injectable/insertable hormonal birth control products. Secondary or barrier forms of contraception include diaphragms, latex condoms, and cervical caps. A diaphragm and cervical cap must each be used with a spermicide.
Estrogen-containing hormonal contraceptives may result in decreased serum valproate levels and potentially lower valproate efficacy. Patients taking DEPAKENE should be advised not to start or stop such products without consulting their physician. Prescribers should monitor valproate serum levels and clinical response when initiating or discontinuing estrogen-containing products (see 7.1.1 Pregnant Women, Female children/Women of childbearing potential/Pregnancy; and 9.4 DrugDrug Interactions, Table 3).
Annual Treatment Reviews by a Specialist
The specialist should, at least annually, review whether valproate is the most suitable treatment option for the patient. The specialist should discuss the Annual Risk Acknowledgement Form, at initiation of treatment and during each annual review and ensure that the patient has understood its content.
Pregnancy Planning
A specialist experienced in the management of epilepsy, must reassess valproate therapy and consider alternative treatment options. Every effort should be made to switch to appropriate alternative treatment prior to conception, and before contraception is discontinued (see 7.1.1 Pregnant Women). If switching is not possible, the woman should receive further counselling regarding valproate risks for the unborn child to support her informed decision making regarding family planning.
In Case of Pregnancy
If a woman receiving valproate treatment becomes pregnant, she must be immediately referred to a specialist to re-evaluate treatment with valproate and consider alternative options. Patients with a valproate exposed pregnancy and their partners should be referred to a specialist experienced in pre-natal medicine for evaluation and counselling regarding the exposed pregnancy (see 7.1.1 Pregnant Women).
Where available, prenatal diagnostic testing to detect neural tube and other defects, should be offered to pregnant women receiving DEPAKENE treatment.
Educational Materials
In order to assist healthcare professionals and patients in avoiding exposure to valproate during pregnancy, the Marketing Authorisation Holder has provided educational materials to reinforce related warnings and provide guidance regarding use of valproate in women of childbearing potential and details of the Pregnancy Prevention Program. A patient guide and patient card should be provided to all women of childbearing potential receiving valproate treatment.
A Risk Acknowledgement Form must be used at the time of treatment initiation and during each annual review of valproate treatment by the specialist, and when a woman is planning a pregnancy or has become pregnant. Specialist should reassess benefits and risks of valproate treatment and determine if the patient should continue to receive valproate therapy.
General
Antiepileptic drugs (AEDs), including DEPAKENE (valproic acid), should be withdrawn gradually to minimize the potential for seizures or increased seizure frequency (see 4.2 Recommended Dose and Dosage Adjustment).
Interaction with Carbapenem Antibiotics
Carbapenem antibiotics (ertapenem, imipenem, meropenem, doripenem) can reduce serum valproic acid concentrations to sub-therapeutic levels. This can result in loss of seizure control in epileptic patients or loss of efficacy in non-epileptics. In some cases of co-administration in epileptic patients, breakthrough seizures have occurred. Increasing valproic acid dose may not be sufficient to overcome this interaction. If co-administration is essential, serum valproic acid concentrations should be monitored daily after initiating carbapenem therapy. Alternative antibacterial or anticonvulsant therapy should be considered if serum valproic acid concentrations drop significantly or seizure control deteriorates (see 9.4 Drug-Drug Interactions, Table 3).
Effects of Valproate on HIV and CMV Viruses Replication
There are in vitro studies that suggest valproate stimulates the replication of the HIV (Human Immunodeficiency Virus) and CMV (Cytomegalovirus) viruses under certain experimental conditions. The clinical relevance of these in vitro data is unknown. Additionally, the relevance of these in vitro findings is uncertain for patients receiving maximally suppressive antiretroviral therapy. Nevertheless, these data should be borne in mind when interpreting the results from regular monitoring of the viral load in HIV infected patients receiving valproate or when following CMV infected patients clinically.
Carcinogenesis and Mutagenesis
Long-term animal toxicity studies indicate that valproic acid is a weak carcinogen or promoter in rats and mice. The significance of these findings for humans is unknown at present (see 16 NON-CLINICAL TOXICOLOGY: Carcinogenicity; and Genotoxicity).
Driving and Operating Machinery
DEPAKENE may produce central nervous system (CNS) depression, especially when combined with another CNS depressant, such as alcohol. Therefore, patients should be advised not to engage in hazardous occupations, such as driving a car or operating dangerous machinery, until it is known that they do not become drowsy from the drug.
Endocrine and Metabolism
Urea Cycle Disorders
DEPAKENE (valproic acid) is contraindicated in patients with known urea cycle disorders. Hyperammonemic encephalopathy, sometimes fatal, has been reported following initiation of DEPAKENE in patients with urea cycle disorders, a group of uncommon genetic abnormalities, particularly ornithine transcarbamylase deficiency. Prior to initiation of DEPAKENE, evaluation for urea cycle disorders (UCD) should be considered in the following patients:
- those with a history of unexplained encephalopathy or coma, encephalopathy associated with protein load, pregnancy-related or postpartum encephalopathy, unexplained mental retardation, or history of elevated plasma ammonia or glutamine;
- those with signs and symptoms of UCD, for example, cyclical vomiting and lethargy, episodic extreme irritability, ataxia, low blood urea nitrogen (BUN), protein avoidance;
- those with a family history of UCD or a family history of unexplained infant deaths (particularly males);
- those with other signs or symptoms of UCD. Patients receiving DEPAKENE who develop symptoms of unexplained hyperammonemic encephalopathy should receive prompt treatment (including discontinuation of DEPAKENE) and be evaluated for underlying urea cycle disorders (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism: Hyperammonemia; and Hyperammonemia and Encephalopathy Associated with Concomitant use of Topiramate, Acetazolamide, Phenobarbital or Phenytoin).
Hyperammonemia
Hyperammonemia has been reported in association with DEPAKENE and may be present despite normal liver function tests. In patients who develop unexplained lethargy and vomiting or changes in mental status, hyperammonemic encephalopathy should be considered as a possible cause and serum ammonia level should be measured. Hyperammonemia should also be considered in patients with hypothermia (see 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism, Hypothermia). If serum ammonia is increased, DEPAKENE should be discontinued. Appropriate interventions for treatment of hyperammonemia should be initiated, and such patients should undergo investigation for underlying urea cycle disorders (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism: Urea Cycle Disorders; and Hyperammonemia and Encephalopathy Associated with Concomitant use of Topiramate, Acetazolamide, Phenobarbital or Phenytoin).
Asymptomatic elevations of serum ammonia are more common and, when present, require close monitoring of serum ammonia levels. If the elevation persists, discontinuation of DEPAKENE should be considered.
Hyperammonemia and Encephalopathy Associated with Concomitant use of Topiramate, Acetazolamide, Phenobarbital or Phenytoin
Concomitant administration of topiramate, acetazolamide, phenobarbital or phenytoin and DEPAKENE has been associated with hyperammonemia with or without encephalopathy in patients who have tolerated either drug alone. Clinical symptoms of hyperammonemic encephalopathy often include acute alterations in level of consciousness and/or cognitive function with lethargy or vomiting. Hypothermia can also be a manifestation of hyperammonemia (see 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism, Hypothermia). In most cases, symptoms and signs abated with discontinuation of either drug. This adverse event is not due to a pharmacokinetic interaction.
It is not known if topiramate, acetazolamide, phenobarbital or phenytoin monotherapy is associated with hyperammonemia.
Patients with inborn errors of metabolism or reduced hepatic mitochondrial activity may be at an increased risk for hyperammonemia with or without encephalopathy. Although not studied, an interaction of topiramate, acetazolamide, phenobarbital or phenytoin and DEPAKENE may exacerbate existing defects or unmask deficiencies in susceptible persons (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism: Urea Cycle Disorders; and Hyperammonemia).
Hypothermia
Hypothermia, defined as an unintentional drop in core body temperature to <35°C (95°F), has been reported in association with DEPAKENE both in conjunction with and in the absence of hyperammonemia. This adverse reaction can also occur in patients using concomitant topiramate with DEPAKENE after starting topiramate treatment or after increasing the daily dose of topiramate (see 9.4 Drug-Drug Interactions, Table 3). Hypothermia may be manifested by a variety of clinical abnormalities including, lethargy, confusion, coma and significant alterations in other major organ systems such as the cardiovascular and respiratory systems. Clinical management and assessment should include examination of blood ammonia levels. Consideration should be given to stopping DEPAKENE in patients who develop hypothermia (see 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism, Hyperammonemia).
Sucrose or Fructose Intolerance
DEPAKENE oral solution contains sucrose, which may be harmful to the teeth. Patients with rare hereditary problems of fructose intolerance, glucose-galactosemalabsorption or sucraseisomaltase insufficiency should not take this medicine.
When prescribing to diabetic patients, the sucrose content should be taken into account (see 6 DOSAGE FORMS, STRENGTHS, COMPOSITION AND PACKAGING).
DEPAKENE oral solution contains sorbitol. Patients with rare hereditary problems of fructose intolerance should not take this medicine.
Hematologic
Thrombocytopenia
Because of reports of thrombocytopenia and inhibition of the second phase of platelet aggregation, and abnormal coagulation parameters (e.g., low fibrinogen), platelet counts and coagulation tests are recommended before initiating therapy and at periodic intervals. It is recommended that patients receiving DEPAKENE be monitored for platelet count and coagulation parameters prior to planned surgery. Clinical evidence of hemorrhage, bruising or a disorder of hemostasis/coagulation is an indication for reduction of DEPAKENE dosage or withdrawal of therapy (see also 7 WARNINGS AND PRECAUTIONS, Hematologic, Dosing-related Adverse Reactions: Thrombocytopenia).
In addition, the findings from a crossover clinical trial conducted with EPIVAL ER (divalproex sodium extended-release tablets), in 44 epilepsy patients, indicate that the frequency of treatment-emergent mild thrombocytopenia (platelet count between 100 to 150 x 109/L) was significantly higher after 12 weeks of treatment with EPIVAL ER than after 12 weeks of treatment with EPIVAL (7 versus 3 low counts, respectively).
Dosing-related Adverse Reactions: Thrombocytopenia
The frequency of adverse effects thrombocytopenia (particularly elevated liver enzymes and thrombocytopenia) may be dose-related. In a clinical trial of EPIVAL (divalproex sodium) as monotherapy in patients with epilepsy, 34/126 patients (27%) rceiving approximately 50 mg/kg/day on average, had at least one value of platelets ≤75 x 109/L. Approximately half of these patients had treatment discontinued with return of platelet counts to normal. In the remaining patients, platelet counts normalized with continued treatment. In this study, the probability of thrombocytopenia appeared to increase significantly at total valproate concentrations of ≥110 mcg/mL (females) or ≥135 mcg/mL (males). The therapeutic benefit which may accompany the higher doses should therefore be weighed against the possibility of a greater incidence of adverse events.
Hepatic / Biliary / Pancreatic
Serious or Fatal Hepatotoxicity
Hepatic failure resulting in fatalities has occurred in patients receiving DEPAKENE and its derivatives. These incidences usually have occurred during the first 6 months of treatment with DEPAKENE. Caution should be observed when administering DEPAKENE to patients with a prior history of hepatic disease. Patients on multiple anticonvulsants, children, those with congenital metabolic disorders, those with severe seizure disorders accompanied by mental retardation, and those with organic brain disease may be at particular risk.
Experience has indicated that children under the age of 2 years are at a considerably increased risk of developing fatal hepatotoxicity, especially those on multiple anticonvulsants, those with congenital metabolic disorders, those with severe seizure disorders accompanied by mental retardation, and those with organic brain disease. The risk in this age group decreased considerably in patients receiving DEPAKENE as monotherapy. Similarly, patients aged 3 to 10 years were at somewhat greater risk if they received multiple anticonvulsants than those who received only DEPAKENE. Above the age of 2 years, experience in epilepsy has indicated that the incidence of fatal hepatotoxicity decreases considerably in progressively older patients. No deaths have been reported in patients over 10 years of age who received DEPAKENE alone.
If DEPAKENE is to be used in children 2 years old or younger, it should be used with extreme caution and as a sole agent. In this patient population, concomitant use of salicylates and DEPAKENE should be avoided due to the risk of liver toxicity. The benefits of therapy should be weighed against the risk (see 7.1.3 Pediatrics).
Serious or fatal hepatotoxicity may be preceded by non-specific symptoms such as loss of seizure control, malaise, weakness, lethargy, facial edema, anorexia, and vomiting. Patients should be monitored closely for appearance of these symptoms. Patients and parents should be instructed to report such symptoms. Because of the non-specific nature of some of the early signs, hepatotoxicity should be suspected in patients who become unwell, other than through obvious cause, while taking DEPAKENE.
Liver function tests should be performed prior to therapy and at frequent intervals thereafter especially during the first 6 months of therapy, especially in patients at risk as described above (see also 9.4 Drug-Drug Interactions, Table 3). However, physicians should not rely totally on serum biochemistry since these tests may not be abnormal in all instances, but should also consider the results of careful interim medical history and physical examination.
In high-risk patients, it might also be useful to monitor serum fibrinogen and albumin for decreases in concentration and serum ammonia for increases in concentration. If changes occur, DEPAKENE should be discontinued. Dosage should be titrated to and maintained at the lowest dose consistent with optimal seizure control.
The drug should be discontinued immediately in the presence of significant hepatic dysfunction, suspected or apparent. In some cases, hepatic dysfunction has progressed in spite of discontinuation of drug. The frequency of adverse effects (particularly elevated liver enzymes and thrombocytopenia) may increase with increasing dose. The therapeutic benefit which may accompany the higher doses should therefore be weighed against the possibility of a greater incidence of adverse effects (see 2 CONTRAINDICATIONS).
Patients with Mitochondrial Disease
Valproate induced acute liver failure and liver-related deaths have been reported in patients with hereditary neurometabolic syndromes caused by mutations in the gene for mitochondrial DNA polymerase γ (POLG) (e.g., Alpers-Huttenlocher Syndrome) at a higher rate than those without these syndromes (see 2 CONTRAINDICATIONS).
POLG-related disorders should be suspected in patients with a family history or suggestive symptoms of a POLG-related disorder, including but not limited to unexplained encephalopathy, refractory epilepsy (focal, myoclonic), status epilepticus at presentation, developmental delays, psychomotor regression, axonal sensorimotor neuropathy, myopathy cerebellar ataxia, opthalmoplegia, or complicated migraine with occipital aura. POLG mutation testing should be performed in accordance with current clinical practice for the diagnostic evaluation of such disorders. The A467T and W748S mutations are present in approximately ⅔ of patients with autosomal recessive POLG-related disorders.
In patients over two years of age who are clinically suspected of having a hereditary mitochondrial disease, DEPAKENE should only be used after other anticonvulsants have failed. This older group of patients should be closely monitored during treatment with DEPAKENE for the development of acute liver injury with regular clinical assessments and liver function test monitoring.
In the presence of significant hepatic dysfunction, suspected or apparent, DEPAKENE should be discontinued and alternative therapy initiated. In some cases, hepatic dysfunction has progressed in spite of discontinuation of the drug (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS).
Pancreatitis
Cases of life-threatening pancreatitis have been reported in both children and adults receiving DEPAKENE. Some of the cases have been described as hemorrhagic with a rapid progression from initial symptoms to death. Some cases have occurred shortly after initial use as well as after several years of use. The rate based upon the reported cases exceeds that expected in the general population and there have been cases in which pancreatitis recurred after rechallenge with DEPAKENE. In clinical trials, there were 2 cases of pancreatitis without alternative etiology in 2,416 patients, representing 1,044 patient-years experience. Patients and guardians should be warned that abdominal pain, nausea, vomiting, and/or anorexia can be symptoms of pancreatitis that require prompt medical evaluation. If pancreatitis is diagnosed, DEPAKENE should ordinarily be discontinued. Alternative treatment for the underlying medical condition should be initiated as clinically indicated.
Monitoring and Laboratory Tests
Since DEPAKENE may interact with concurrently administered drugs which are capable of enzyme induction, periodic plasma concentration determinations of valproate and concomitant drugs are recommended during the early course of therapy and whenever enzyme-inducing drugs are introduced or withdrawn (see 9 DRUG INTERACTIONS).
Monitoring Valproate Concentrations
Protein binding of valproate is reduced in the elderly, in patients with renal impairment, and in the presence of other drugs (e.g., acetylsalicylic acid). Accordingly, measurements of plasma levels of valproate may be misleading in these patients, as actual drug exposure may be higher than measured values(see 7.1.4 Geriatrics; 7 WARNINGS AND PRECAUTIONS: Hepatic/Biliary/Pancreatic; Endocrine and Metabolism, Hyperammonemia; Hematologic, Thrombocytopenia; and 9.4 Drug-Drug Interactions, Table 3).
Musculoskeletal / Rhabdomyolysis
Rare cases of rhabdomyolysis, independent of neuroleptic malignant syndrome, have been reported to occur in patients treated with DEPAKENE. Cases have included renal failure and fatalities.
Patients should be carefully monitored for muscle pain, tenderness or weakness, particularly if accompanied by malaise or fever or tea-coloured urine. Blood creatine phosphokinase (CPK) levels should be assessed in patients experiencing these symptoms and DEPAKENE therapy should be discontinued if markedly elevated CPK levels are measured or if the patient develops signs and symptoms indicative of rhabdomyolysis.
Caution should be exercised in prescribing DEPAKENE to patients with predisposing/risk factors, including: prior history of muscular disorders such as CPT II deficiency (carnitine palmitoyltransferase DEPAKENE, valproic acid Product Monograph Page 18 of 57 type II); uncontrolled hypothyroidism; hepatic or renal impairment; concomitant medications that are known to be associated with rhabdomyolysis (e.g.,statins, antipsychotics, diuretics, some antidepressants).
Neurologic
Brain Atrophy
There have been postmarketing reports of reversible and irreversible cerebral and cerebellar atrophy with neurological symptoms, in children, adults, and the elderly, receiving valproate therapy. A temporal relationship between valproate therapy and the development of cerebral atrophy and associated signs and symptoms was also demonstrated. In some cases, symptoms disappeared after valproate discontinuation but patients recovered with permanent sequelae (see 8 ADVERSE REACTIONS). The motor and cognitive functions of patients on valproate should be routinely monitored and drug should be discontinued in the presence of suspected or apparent signs of brain atrophy.
Neurological Problems in Children after in utero Exposure to Valproate
Reports of cerebral atrophy with various forms of neurological problems including cognitive developmental delays, psychomotor impairment and decreased IQ scores have been reported in children who were exposed in utero to valproate products (see 7.1.1 Pregnant Women).
Aggravated convulsions
As with other antiepileptic drugs, some patients may experience a worsening of convulsion frequency and severity, or the onset of new types of convulsions with valproate. Postmarketing reports of serious aggravated seizures have been reported for valproic acid including status epilepticus and death. In case of aggravated convulsions, patients should be advised to consult their physician immediately.
Psychiatric
Suicidal Behaviour and Ideation
Suicidal ideation and behaviour have been reported in patients treated with antiepileptic agents in several indications.
All patients treated with antiepileptic drugs (AEDs), irrespective of indication, should be monitored for signs of suicidal ideation and behaviour and appropriate treatment should be considered. Patients (and caregivers of patients) should be advised to seek medical advice should signs of suicidal ideation or behaviour emerge.
An FDA meta-analysis of randomized placebo controlled trials, in which AEDs were used for various indications, has shown a small increased risk of suicidal ideation and behaviour in patients treated with these drugs. The mechanism of this risk is not known.
There were 43,892 patients treated in the placebo controlled clinical trials that were included in the meta-analysis. Approximately 75% of patients in these clinical trials were treated for indications other than epilepsy and, for the majority of non-epilepsy indications the treatment (AED or placebo) was administered as monotherapy. Patients with epilepsy represented approximately 25% of the total number of patients treated in the placebo controlled clinical trials and, for the majority of epilepsy patients, treatment (AED or placebo) was administered as adjunct to other antiepileptic agents (i.e., patients in both treatment arms were being treated with one or more AED). Therefore, the small increased risk of suicidal ideation and behaviour reported from the meta-analysis (0.43% for patients on AEDs compared to 0.24% for patients on placebo) is based largely on patients that received monotherapy treatment (AED or placebo) for non-epilepsy indications. The study design does not allow an estimation of the risk of suicidal ideation and behaviour for patients with epilepsy that are taking AEDs, due both to this population being the minority in the study, and the drug-placebo comparison in this population being confounded by the presence of adjunct AED treatment in both arms.
Behavioural Disorders
There have been postmarketing reports of behavioural disorders, including aggression, agitation, abnormal behaviour, psychomotor hyperactivity, disturbance in attention, and learning disorders. Although patients of all ages were affected, including the elderly, a large number of cases were reported in children (see 8.1 Adverse Reaction Overview, Pediatric Population). There was no clear trend with respect to valproate dose. In some cases, patients improved or recovered following valproate discontinuation. Attention deficit hyperactivity disorder (ADHD), Autism spectrum disorders and developmental delay have been reported from in utero exposure (see 7.1.1 Pregnant Women).
Renal
Renal Impairment
Renal impairment is associated with an increase in the unbound fraction of valproate. In several studies, the unbound fraction of valproate in plasma from renally impaired patients was approximately double that for subjects with normal renal function. Accordingly, monitoring of total concentrations in patients with renal impairment may be misleading since free concentrations may be substantially elevated whereas total concentrations may appear to be normal. Hemodialysis in renally impaired patients may remove up to 20% of the circulating valproate.
Reproductive Health: Female and Male Potential
Fertility
Amenorrhea, polycystic ovaries and increased testosterone levels have been reported in women using valproate.
The effect of DEPAKENE on the development of the testis in humans is unknown (see 16 NONCLINICAL TOXICOLOGY, Reproductive and Developmental Toxicology, Fertility for results in animal studies). Valproate administration has been associated with reduced semen quality in humans and thus may impair fertility in men (see 8 ADVERSE REACTIONS). Discontinuation or dose reduction of valproate may be associated with the improvement of impaired male fertility markers and could be linked with successful conception, as observed in some case reports.
Sensitivity / Resistance
Multi-organ Hypersensitivity Reaction
Multi-organ hypersensitivity reactions have been rarely reported in close temporal association to the initiation of DEPAKENE in adult and pediatric patients (median time to detection 21 days; range 1 to 40). Although there have been a limited number of reports, many of these cases resulted in hospitalization and at least one death has been reported. Signs and symptoms of this disorder were diverse; however, patients typically, although not exclusively, presented with fever and rash associated with other organ system involvement. Other associated manifestations may include lymphadenopathy, hepatitis, liver function test abnormalities, hematological abnormalities (e.g., eosinophilia, thrombocytopenia, neutropenia), pruritus, nephritis, oliguria, hepato-renal syndrome, arthralgia, and asthenia. Because the disorder is variable in its expression, other organ system symptoms and signs, not noted here may occur. If this reaction is suspected, DEPAKENE should be discontinued and an alternative treatment started. Although the existence of cross sensitivity with other drugs that produce this syndrome is unclear, the experience amongst drugs associated with multi-organ hypersensitivity would indicate this to be a possibility.
Skin
Serious Skin Reactions
The dose of lamotrigine should be reduced when co-administered with DEPAKENE. Serious skin reactions (such as Stevens-Johnson syndrome and Toxic Epidermal Necrolysis) have been reported with concomitant lamotrigine and DEPAKENE administration (see Lamotrigine Product Monograph for details on lamotrigine dosing with concomitant DEPAKENE administration).
7.1 Special Populations
7.1.1 Pregnant Women
Female children/Women of childbearing potential/Pregnancy
DEPAKENE can cause fetal harm when administered to pregnant women. DEPAKENE use during pregnancy is associated with an increased risk of severe birth defects such as neural tube defects (e.g., spina-bifida), craniofacial defects, cleft palate, cardiovascular malformations (e.g., atrial septal defect), hypospadias, etc. In some cases, fatal outcomes have been reported (see 7.1.1 Pregnant Women, Birth Defects).
Valproate is contraindicated as treatment for epilepsy during pregnancy unless there is no suitable alternative treatment (see 2 CONTRAINDICATIONS).
DEPAKENE should not be used to treat female children or women of childbearing potential unless alternative treatments are ineffective or not toleratedand unless the conditions of the Pregnancy Prevention Program are fulfilled (see 7.1.1 Pregnant Women, Pregnancy Prevention Programbelow). The benefit and risk should be carefully re-assessed, at least annually, at puberty, and urgently when a woman of childbearing potential plans a pregnancy or becomes pregnant. Since some of the congenital malformations occur in the first trimester of pregnancy before many women know that they are pregnant, all women of childbearing potential should be informed of the potential hazard to the fetus from exposure to DEPAKENE. Women of childbearing potential must use at least one effective and reliable method of contraception (preferably a user-independent form) or two complementary forms of contraception during treatment with DEPAKENE (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program).
Valproate does not reduce the efficacy of hormonal contraceptives. However, estrogen-containing products, including estrogen-containing hormonal contraceptives, may increase the clearance of valproate, which may result in decreased serum concentration of valproate and potentially increased seizure frequency. Patients taking DEPAKENE should be advised not to start or stop estrogen-containing products (including oral contraceptives) without consulting their physician. Prescribers should monitor valproate plasma levels and clinical response (seizure control) when initiating, or discontinuing estrogen-containing products (see 9.4 Drug-Drug Interactions, Table 3).
Pregnancy Exposure Risk Related to Valproate
Both valproate adjunctive and monotherapy are frequently associated with abnormal pregnancy outcomes. Available data show an increased risk of major congenital malformations and neurodevelopmental disorders in both valproate monotherapy and polytherapy compared to the population not exposed to valproate.
Tests to detect neural tube and other defects using current accepted procedures should be considered as part of routine prenatal care in pregnant women receiving DEPAKENE.
Pregnancy Registry
Pregnant patients taking DEPAKENE should be encouraged to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. This can be done by calling the toll free number 1-888-233-2334, and must be done by patients themselves. Information on the registry can also be found at the following website: http://www.aedpregnancyregistry.org/.
Pregnancy Prevention Program
Information on the Pregnancy Prevention Program including educational resources, as well as to report suspected embryo-fetal exposure to valproate, can be found at the following website: www.depakene.ca.
If a woman is planning a pregnancy:
A specialist experienced in the management of epilepsy, must reassess valproate therapy and consider alternative treatment options. Every effort should be made to switch to appropriate alternative treatment prior to conception, and before contraception is discontinued (see 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program). If switching is not possible, the woman should receive further counselling regarding the valproate risks for the unborn child to support her informed decision making regarding family planning.
Pregnant Women
Valproate as treatment for epilepsy is contraindicated in pregnancy unless there is no suitable alternative treatment (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program).
If a woman using valproate becomes pregnant, she must be immediately referred to a specialist to consider alternative treatment options.
During pregnancy, maternal tonic-clonic seizures and status epilepticus with hypoxia may carry a particular risk of death for mother and the unborn child. If, despite the known risks of valproate in pregnancy and after careful consideration of alternative treatment, in exceptional circumstances, a pregnant woman must receive valproate for epilepsy, it is recommended to:
- Use the lowest effective dose and divide the daily dose of valproate into several small doses to be taken throughout the day;
- Consider the use of a prolonged release formulation of DEPAKENE, which may be preferable to immediate-release formulations in order to avoid high peak plasma concentrations.
All patients with a valproate exposed pregnancy and their partners should be referred to a specialist experienced in pre-natal medicine for evaluation and counselling. Specialized prenatal monitoring should take place to detect possible occurrence of neural tube defects or other malformations. Folate supplementation (5 mg daily) before the pregnancy may decrease the risk of neural tube defects which may occur in all pregnancies. However, available evidence does not suggest that folate can prevent birth defects or malformations due to valproate exposure.
Valproate is contraindicated as treatment for epilepsy during pregnancy unless there is no suitable alternative to treat epilepsy. Valproate is contraindicated for use in women of childbearing potential unless the conditions of the Pregnancy Prevention Program are fulfilled (see 2 CONTRAINDICATIONS and 7 WARNINGS AND PRECAUTIONS, Pregnancy Prevention Program).
Risk in the neonate
Cases of hemorrhagic syndrome have been reported very rarely in neonates whose mothers have taken valproate during pregnancy. This hemorrhagic syndrome is related to thrombocytopenia, hypofibrinogenemia and/or to a decrease in other coagulation factors.
Afibrinogenemia has also been reported and can be fatal. However, this syndrome must be distinguished from the decrease of vitamin-K factors induced by phenobarbital and other enzymes. Therefore, in neonates, platelet count, plasma levels of fibrinogen, coagulation tests and coagulation factors should be investigated.
Cases of hypoglycemia have been reported in neonates whose mothers have taken valproate during the third trimester of pregnancy.
Cases of hypothyroidism have been reported in neonates whose mothers have taken valproate during pregnancy.
Withdrawal syndrome (symptoms include: agitation, irritability, hyperexcitability, jitteriness, hyperkinesia, tonicity disorders, tremor, convulsions and feeding disorders) may occur, in the days following birth, in neonates whose mothers have taken valproate during the last trimester of pregnancy.
Birth Defects
Summary:
- Valproate can cause fetal harm when administered to pregnant women;
- Maternal valproate use can cause neural tube defects (e.g., spina-bifida) and other structural abnormalities (e.g., craniofacial defects, cardiovascular malformations such as atrial septal defect, hypospadias, limb malformations such as club foot and polydactyly);
- The rate of congenital malformations among babies born to mothers using valproate monotherapy is about four times higher than the rate among babies born to epileptic mothers using other anti-epileptic monotherapies.
- The risk of major congenital malformations in children after in utero exposure to anti-epileptic polytherapy including valproate is higher than that of anti-epileptic drug polytherapy not including valproate.
- This risk is dose-dependent in valproate monotherapy, and available data suggest it is valproate dose-dependent in polytherapy. However, a threshold dose below which no risk exists cannot be established.
Data:
Data described below were gained almost exclusively from women who received valproate to treat epilepsy. Data from Pregnancy Registries indicate an increased risk of congenital anomalies in infants exposed to DEPAKENE monotherapy during the first trimester of pregnancy as compared to other antiepileptic drugs. Based on Pregnancy Registry data and the United States Centers for Disease Control (CDC), the estimated risk of valproate-exposed women having children with spinabifida, oral clefts, neural tube defects, and hypospadias is approximately 1 to 2% as compared to the risk of spina-bifida in the general population which is about 0.06 to 0.07%.
In a study using NAAED Pregnancy Registry data, 16 cases of major malformations following prenatal valproate exposure were reported among offspring of 149 enrolled women who used valproate during pregnancy. Three of the 16 cases were neural tube defects; the remaining cases included craniofacial defects, cardiovascular malformations and malformations of varying severity involving other body systems. The NAAED Pregnancy Registry has reported a major malformation rate of 10.7% in the offspring of women exposed to valproate monotherapy during pregnancy (average daily dose: 1000 mg; range: 500 – 2000 mg/day) as compared to major malformation rate of 2.9% among 1,048 epileptic women who received any other antiepileptic drug monotherapy during pregnancy. These data show a four-fold increased risk for any major malformation following valproate exposure in utero compared to the risk following exposure in utero to any other antiepileptic drug monotherapy.
Data derived from a meta-analysis (including registries and cohort studies) has shown that 10.93% of children of epileptic women exposed to valproate monotherapy during pregnancy suffer from major congenital malformations (95% CI 8.91-13.13). This is greaterthan the risk of major malformations in the general population (about 2-3%).
Available data show an increased incidence of minor and major malformations. The most common types of malformations include neural tube defects, facial dysmorphism, cleft lip and palate, craniostenosis, cardiac, renal and urogenital defects, limb defects (including bilateral aplasia of the radius), and multiple anomalies involving various body systems.
In utero exposure to valproate may result in eye malformations that have been observed in conjunction with other congenital malformations. In most cases, valproate was taken as monotherapy during the whole pregnancy and not a specific trimester.
In utero exposure to various therapeutic doses of valproate, during any trimester of pregnancy, can also result in hearing impairment or deafness due to ear and/or nose malformations (secondary effect) and/or to direct toxicity on the hearing function. All cases to date were reported as serious and included both unilateral and bilateral deafness or hearing impairment. When outcomes were reported, the majority of the cases remained unresolved. In 58% of the cases, the age of diagnosis of hearing impairment or deafness was within the first 4 weeks following birth. Monitoring of signs and symptoms of ototoxicity is recommended.
Risk of Neurological Problems from in utero Exposure
Cerebral Atrophy
Exposure in utero to valproate products has been associated with cerebral atrophy with varying degrees/manifestations of neurological compromise, including developmental delays and psychomotor impairment (see 8 ADVERSE REACTIONS and 7 WARNINGS AND PRECAUTIONS, Neurologic, Brain Atrophy).
Neurodevelopmental Disorders
Available data show that exposure to valproate in utero can have adverse effects on mental and physical development of the exposed children. Whether a child's in utero exposure to valproate is as monotherapy, or as polytherapy with other anti-epileptic drugs, the risks of neurodevelopmental disorders are significantly greater than for children in the general population or those born to untreated epileptic mothers. The risks seem to be dose-dependent but a threshold dose below which no risk exists, cannot be established. The exact gestational period for risk of these effects is uncertain and it is possible that the risk exists throughout the entire pregnancy.
Studies in preschool children exposed to valproate monotherapy in utero show that up to 30-40% experience delays in their early development such as talking and walking later, lower intellectual abilities, poor language skills (speaking and understanding) and memory problems.
Decreased IQ
Valproate can cause decreased Intelligence Quotient (IQ) scores in children following in utero exposure. Although it is not known exactly when during pregnancy cognitive effects in valproateexposed children occur, there is a risk that it may occur early in pregnancy.
IQ measured in school aged children (age 6) with a history of valproate exposure in uterowas on average 7-10 points lower than those children exposed to other antiepileptics. There is evidence in children exposed to valproate that the risk of intellectual impairment may be independent of maternal IQ.
There are limited data on long term outcomes.
Autism and/or Autism Spectrum Disorders
A population-based study was conducted in Denmark based on various national patient registries including the Danish Medical Birth Register. This study showed that children exposed to valproate in utero are at increased risk of autism spectrum disorders (approximately 3-fold) and childhood autism (approximately 5-fold) compared to the children of unexposed epileptic women in the same study.
Attention Deficit Hyperactivity Disorder (ADHD)
Another population-based study in Denmark was also conducted based on various national patient registries including the Danish Medical Birth Register. This study showed that children exposed to valproate in utero are at increased risk of developing ADHD compared to the children of unexposed epileptic women in the same study. Data show that 8.4% of the children exposed to valproate in uterowere diagnosed with ADHD compared to 3.2% of unexposed children in the same study.
Although available studies have some limitations, the weight of the evidence supports a causal association between valproate exposure in utero and subsequent adverse effects on neurodevelopment, including increases in the occurrence of autism spectrum disorders.
Coagulation Abnormalities
There have been reported postmarketing cases of coagulation abnormalities in patients of all ages receiving valproate therapy. These include thrombocytopenia, hypofibrinogenemia, and/or decrease in other coagulation factors, which can lead to bleeding and other complications, especially in the cases of decrease in factors VII, VIII and XIII. These abnormalities may not necessarily be dose-dependent. Some of the hemorrhage manifestations may include mucosal bleeding (e.g., menorrhagia, epistaxis, hematuria, melena), easy-bruising, soft-tissue hematoma, hemarthrosis, and intracranial hemorrhage. Caution should be taken in patients taking valproate and anticoagulants and in cases of injury or surgery to avoid life-threatening or fatal bleeding (see 7 WARNINGS AND PRECAUTIONS, Hematologic, Thrombocytopenia).
Pregnant women taking DEPAKENE may also develop coagulation abnormalities, which may result in hemorrhagic complications in the neonate including death (see 7 WARNINGS AND PRECAUTIONS, Hematologic, Thrombocytopenia). If DEPAKENE is used in pregnancy, the coagulation parameters should be monitored carefully.
Hepatic Failure
Hepatic failure, resulting in the death of a newborn and of an infant has been reported following the use of valproate during pregnancy.
Hypoglycemia
Serious postmarketing reports of hypoglycemia have been received for neonates whose mothers received DEPAKENE treatment during pregnancy. In most cases, DEPAKENE was the only reported antiepileptic drug (AED). Most of these neonates also displayed other congenital anomalies including hypospadias, complex facial dysmorphism, limb anomalies, severe cardiac anomalies, etc. Therefore, if a decision has been made to use DEPAKENE during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be made aware of the potential hazard to the fetus.
Thyroid Gland Abnormalities
Cases of hypothyroidism have been reported in neonates whose mothers have taken valproate during pregnancy. There have also been reported cases of increased serum thyroid stimulating hormone or decreased serum thyroxine levels in children receiving valproate therapy. In addition, there have been reported cases of hypothyroidism and hyperthyroidism in adults and children receiving valproate monotherapy.
Teratogenicity in Animals
Animal studies have demonstrated valproic acid induced teratogenicity (see 16 NON-CLINICAL TOXICOLOGY, Reproductive and Developmental Toxicology), and studies in human females have demonstrated placental transfer of the drug. Increased frequencies of malformations, as well as intrauterine growth retardation and death, have been observed in mice, rats, rabbits, and monkeys following prenatal exposure to valproate. Malformations of the skeletal system are the most common structural abnormalities produced in experimental animals, but neural tube closure defects have been seen in mice exposed to maternal plasma valproate concentrations exceeding 230 mcg/mL (2.3 times the upper limit of the human therapeutic range for epilepsy) during susceptible periods of embryonic development.
Administration of an oral dose of 200 mg/kg/day or greater (50% of the maximum human daily dose or greater on a mg/m² basis) to pregnant rats during organogenesis produced malformations (skeletal, cardiac and urogenital) and growth retardation in the offspring. These doses resulted in peak maternal plasma valproate levels of approximately 340 mcg/mL or greater (3.4 times the upper limit of the human therapeutic range for epilepsy or greater). Behavioural deficits have been reported in the offspring of rats given a dose of 200 mg/kg/day throughout most of pregnancy.
An oral dose of 350 mg/kg/day (approximately 2 times the maximum human daily dose on a mg/m² basis) produced skeletal and visceral malformations in rabbits exposed during organogenesis. Skeletal malformations, growth retardation, and death were observed in rhesus monkeys following administration of an oral dose of 200 mg/kg/day (equal to the maximum human daily dose on a mg/m² basis) during organogenesis. This dose resulted in peak maternal plasma valproate levels of approximately 280 mcg/mL (2.8 times the upper limit of the human therapeutic range for epilepsy).
7.1.2 Breast-feeding
DEPAKENE is secreted in breast milk. Concentrations in breast milk have been reported to be 1 to 10% of maternal serum concentrations. Women should not breastfeed during DEPAKENE treatment and for one month after discontinuation of the drug. Based on literature and clinical experience, hematological disorders have been shown in breastfed newborns/infants of treated women.
7.1.3 Pediatrics
Experience has indicated that children under the age of 2 years are at a considerably increased risk of developing fatal hepatotoxicity, especially those on multiple anticonvulsants, those with congenital metabolic disorders, those with severe seizure disorders accompanied by mental retardation, and those with organic brain disease. When DEPAKENE is used in patients 2 years of age or younger, it should be used with extreme caution and as a sole agent, and the concomitant use of salicylates should be avoided due to the risk of liver toxicity. The benefits of therapy should be weighed against the risks. See 2 CONTRAINDICATIONS; and 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Serious or Fatal Hepatotoxicity.
Above the age of 2 years, experience in epilepsy has indicated that the incidence of fatal hepatotoxicity decreases considerably in progressively older patient groups.
In patients over two years of age who are clinically suspected of having a hereditary mitochondrial disease, DEPAKENE should only be used after other anticonvulsants have failed. (see 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Patients with Mitochondrial Disease).
Younger children, especially those receiving enzyme-inducing drugs, will require larger maintenance doses to attain targeted total and unbound valproate concentrations. The variability in free fraction limits the clinical usefulness of monitoring total serum valproate concentrations. Interpretation of valproate concentrations in children should include consideration of factors that affect hepatic metabolism and protein binding.
7.1.4 Geriatrics
Alterations in the kinetics of unbound valproate in the elderly (≥65 years of age) indicate that the initial dosage should be reduced in this population (see 4 DOSAGE AND ADMINISTRATION and 10.3 Pharmacokinetics, Special Populations and Conditions, Geriatrics).
The safety and efficacy of DEPAKENE in elderly patients with epilepsy has not been evaluated in clinical trials. Caution should thus be exercised in dose selection for an elderly patient, recognizing the more frequent hepatic and renal dysfunctions, and limited experience with DEPAKENE in this population.
A study of elderly patients revealed valproate-related somnolence and discontinuation of DEPAKENE for this adverse event (see 7.1.3 Geriatrics, Somnolence in the Elderly). The starting dose should be reduced in elderly patients, and dosage reductions or discontinuation should be considered in patients with excessive somnolence (see 4 DOSAGE AND ADMINISTRATION).
Somnolence in the elderly
In a group of elderly patients (mean age 83 years old, n = 172), DEPAKENE doses were increased by 125 mg/day to a target dose of 20 mg/kg/day. Compared to placebo a significantly higher number of valproate-treated patients had somnolence, and although not statistically significant, a higher number of valproate-treated patients experienced dehydration. Discontinuations for somnolence were also significantly higher in valproate-treated patients compared to placebo. In approximately one-half of the patients with somnolence, there was also associated reduced nutritional intake and weight loss. In elderly patients, dosage should be increased more slowly and with regular monitoring for fluid intake, dehydration, somnolence, urinary tract infection and other adverse events. Dose reductions or discontinuation of DEPAKENE should be considered in patients with decreased food or fluid intake and in patients with excessive somnolence (see 4 DOSAGE AND ADMINISTRATION).
Adverse reactions
8.1 Adverse Reaction Overview
The most commonly reported adverse reactions are nausea, vomiting and indigestion. Since DEPAKENE (valproic acid) has usually been used with other antiepileptics, it is not possible in most cases to determine whether the adverse reactions mentioned in this section are due to DEPAKENE alone or to the combination of drugs.
Adverse events that have been reported with DEPAKENE from epilepsy trials, spontaneous reports, and other sources are listed below by system organ class.
Blood and Lymphatic System Disorders
Thrombocytopenia and inhibition of the secondary phase of platelet aggregation may be reflected in altered bleeding time, petechiae, bruising, hematoma formation, epistaxis, and hemorrhage (see 7 WARNINGS AND PRECAUTIONS, Hematologic, Thrombocytopenia). Relative lymphocytosis, macrocytosis and hypofibrinogenemia have been noted. Leukopenia and eosinophilia have also been reported. Anemia, including macrocytic with or without folate deficiency, aplastic anemia, pancytopenia, bone marrow suppression, agranulocytosis and acute intermittent porphyria have been reported.
Cardiac Disorders
Bradycardia.
Ear and Labyrinth Disorders
Hearing loss, either reversible or irreversible, has been reported; however, a cause and effect relationship has not been established. Ear pain has also been reported.
Gastrointestinal Disorders
Nausea, vomiting and indigestion are the most commonly reported side effects at the initiation of therapy. These effects are usually transient and rarely require discontinuation of therapy. Diarrhea, abdominal cramps, constipation and gingival disorder (mainly gingival hyperplasia) have also been reported. There have been reports of acute pancreatitis, including rare fatal cases, occurring in association with DEPAKENE therapy (see 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Pancreatitis). Parotid gland swelling has also been reported in patients receiving DEPAKENE.
General Disorders and Administration Site Conditions
Edema of the extremities, fever and hypothermia
Hepatobiliary Disorders
Minor elevations of transaminases [e.g., serum glutamic-oxaloacetic transaminase (SGOT) and serum glutamic-pyruvic transaminase (SGPT)] and lactate dehydrogenase (LDH) are frequent and appear to be dose-related. Occasionally, laboratory tests also show increases in serum bilirubin and abnormal changes in other liver function tests. These results may reflect potentially serious hepatotoxicity (see 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Serious or Fatal Hepatotoxicity).
Immune System Disorder
Allergic reaction, anaphylaxis
Infections and Infestations
Pneumonia and otitis media
Investigations
Abnormal thyroid function tests (including both hyperthyroidism and hypothyroidism) (see 7.1.1 Pregnant Women, Thyroid Gland Abnormalities and 9.7 Drug-Laboratory Test Interactions).
Metabolism and Nutrition Disorders
Hyperammonemia (see 7 WARNINGS AND PRECAUTIONS, Endocrine and Metabolism, Hyperammonemia), hyponatremia, biotin deficiency/biotinidase deficiency and inappropriate antidiuretic hormone (ADH) secretion. There have been rare reports of Fanconi syndrome (proximal renal tubular dysfunction) occurring primarily in children. Decreased carnitine concentrations have been reported although the clinical relevance is undetermined. Hyperglycinemia has been reported and associated with a fatal outcome in patient with pre-existing nonketotic hyperglycinemia.
Anorexia with some weight loss and increased appetite with some weight gain have also been reported.
Obesity has been reported in post-marketing experience.
Musculoskeletal and Connective Tissue Disorders
Weakness, rhabdomyolysis and bone pain have been reported (see 7 WARNINGS AND PRECAUTIONS, Musculoskeletal/Rhabdomyolysis).
Reports have been received of decreased bone mass, potentially leading to osteoporosis and osteopenia, during long-term therapy with some anticonvulsant medications, including DEPAKENE. Some studies have indicated that supplemental calcium and vitamin D may be of benefit to patients who are on chronic DEPAKENE therapy.
A lupus erythematosus-like syndrome has been reported rarely.
Neoplasms Benign, Malignant and Unspecified (including cysts and polyps)
Myelodysplastic syndrome in both adults and children (all children were on valproate monotherapy). In some cases in adults and/or children, myelodysplastic syndrome was reversible upon valproate discontinuation.
Nervous System Disorders
Sedative effects have been noted in patients receiving DEPAKENE alone but occur most often in patients on combination therapy. Sedation usually disappears upon reduction of other anti-epileptic medication.
Hallucination, ataxia, headache, nystagmus, diplopia, asterixis, "spots before the eyes", tremor (may be dose-related), confusion, dysarthria, dizziness, hypesthesia, vertigo, incoordination, memory impairment, cognitive disorder, and extrapyramidal disorders including parkinsonism have been reported with the use of valproate. Rare cases of coma have been reported in patients receiving DEPAKENE alone or in conjunction with phenobarbital.
Encephalopathy, with or without fever or hyperammonemia, has been reported without evidence of hepatic dysfunction or inappropriate valproate plasma levels. Most patients recovered, with noted improvement of symptoms, upon discontinuation of the drug.
There have been postmarketing reports of reversible and irreversible cerebral and cerebellar atrophy associated with the use of valproate products. In some cases the patients recovered with permanent sequelae (see 7 WARNINGS AND PRECAUTIONS, Neurologic, Brain Atrophy). Cerebral atrophy seen in children exposed to valproate in utero led to various forms of neurological events, including developmental delays and psychomotor impairment.
Congenital malformations and developmental disorders have also been reported. See 7.1.1 Pregnant Women, Pregnancy Exposure Risk Related to Valproate, - Birth Defects; and - Risk of Neurological Problems from in utero Exposure.
Aggravated convulsions (increase in number of seizures or appearance of new seizure type or worsening of seizures) have been reported in patients with epilepsy treated with valproate monotherapy.
Psychiatric disorders
Emotional upset, depression, psychosis, aggression, psychomotor hyperactivity, hostility, agitation, disturbance in attention, abnormal behaviour, learning disorder and behavioural deterioration (see 7 WARNINGS AND PRECAUTIONS, Psychiatric).
Renal and Urinary Disorders
Enuresis, urinary incontinence, acute renal failure, tubulointerstitial nephritis and urinary tract infection.
Reproductive System and Breast Disorders
There have been reports of irregular menses, secondary amenorrhea, breast enlargement and galactorrhea in patients receiving DEPAKENE. Hyperandrogenism (hirsutism, virilism, acne, male pattern alopecia, and/or androgen increased).
There have been post-marketing reports of aspermia, azoospermia, decreased sperm count, decreased spermatozoa motility, abnormal spermatozoa morphology and ultimately infertility in male patients who received sodium valproate products (effects may be improved by dose reduction or discontinuation).
There have been rare spontaneous reports of polycystic ovary disease. A cause and effect relationship has not been established.
Respiratory, Thoracic and Mediastinal Disorders
Increased cough, pleural effusion
Skin and Subcutaneous Tissue Disorders
Transient and/or dose related alopecia (hair loss), hair disorders (such as hair texture abnormal, hair colour changes, hair growth abnormal), have been observed. Skin rash, photosensitivity, generalized pruritus, erythema multiforme, Stevens-Johnson syndrome, and petechiae have rarely been noted.
Rare cases of Toxic Epidermal Necrolysis (TEN) have been reported including a fatal case of a 6 month old infant taking DEPAKENE and several other concomitant medications. An additional case of Toxic Epidermal Necrolysis resulting in death was reported in a 35 year old patient with AIDS taking several concomitant medications and with a history of multiple cutaneous drug reactions.
Serious skin reactions have been reported with concomitant administration of lamotrigine and DEPAKENE (see 9.4 Drug-Drug Interactions, Table 3).
Cutaneous vasculitis has also been reported.
Nail and nail bed disorders have also been reported in post-marketing experience.
Pediatric Population
The safety profile of valproate in the pediatric population is comparable to adults, but some adverse reactions are more severe or principally observed in the pediatric population. For example, there is a particular risk of severe liver damage in infants and young children especially under the age of 3 years. These risks appear to decrease with increasing age; young children are also at a higher risk of severe pancreatitis, which may result in fatalities (see 3 SERIOUS WARNINGS AND PRECAUTIONS BOX, Hepatotoxicity, 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic: Serious or Fatal Hepatotoxicity; and Pancreatitis). Psychiatric disorders such as aggression, agitation, disturbance in attention, abnormal behavior, psychomotor hyperactivity and learning disorder are principally observed in the pediatric population (see 7 WARNINGS AND PRECAUTIONS, Psychiatric, Behavioural Disorders).
8.5 Post-Market Adverse Reactions
Adverse Events in Elderly Patients
In elderly patients (above 65 years of age), there were more frequent reports of accidental injury, infection, pain, and to a lesser degree, somnolence and tremor, when compared to patients 18 to 65 years of age. Somnolence and tremor tended to be associated with the discontinuation of DEPAKENE.
Reproductive Findings
There have been very rare reports of testicular atrophy, decreased semen volume, male hypogonadism, decreased blood testosterone and/or increased blood prolactin levels in patients treated with valproate products. There are insufficient data to determine the exact effect of valproate therapy on testicular development in humans (see 16 NON-CLINICAL TOXICOLOGY, Reproductive and Developmental Toxicology, Fertility).
Drug interactions
9.1 Serious Drug Interactions
Serious Drug Interactions
- Rare cases of coma have been reported in patients receiving DEPAKENE alone or in conjunction with phenobarbital (see 9.4 Drug-Drug Interactions, Table 3).
- Serious skin reactions (such as Stevens-Johnson syndrome and Toxic Epidermal Necrolysis) have been reported with concomitant lamotrigine and DEPAKENE administration (see 9.4 Drug-Drug Interactions, Table 3).
9.2 Drug Interactions Overview
DEPAKENE has been found to be a weak inhibitor of some P450 isozymes, epoxide hydrase, and glucuronyl transferases.
Drugs that affect the level of expression of hepatic enzymes, particularly those that elevate levels of glucuronyl transferases (such as ritonavir; see Table 3 below), may increase the clearance of valproate. For example, phenytoin, carbamazepine, and phenobarbital (or primidone) can double the clearance of valproate. Thus, patients on DEPAKENE monotherapy will generally have longer half-lives and higher concentrations than patients receiving polytherapy with antiepilepsy drugs.
In contrast, drugs that are inhibitors of cytochrome P450 isozymes, such as antidepressants, may be expected to have little effect on valproate clearance because cytochrome P450 microsomal mediated oxidation is a relatively minor secondary metabolic pathway compared to glucuronidation and betaoxidation.
The concomitant administration of DEPAKENE with drugs that exhibit extensive protein binding (e.g., acetylsalicylic acid, carbamazepine, dicumarol, warfarin, tolbutamide, and phenytoin) may result in alteration of serum drug levels.
Since DEPAKENE may interact with concurrently administered drugs which are capable of enzyme induction, periodic plasma concentration determinations of valproate and concomitant drugs are recommended during the early course of therapy and whenever enzyme-inducing drugs are introduced or withdrawn.
9.3 Drug-Behavioural Interactions
Refer to 7 WARNINGS AND PRECAUTIONS, Driving and Operating Machinery for details.
9.4 Drug-Drug Interactions
Table 3 provides information about the potential influence of several commonly prescribed medications on DEPAKENE pharmacokinetics as well as the potential influence of DEPAKENE on the pharmacokinetics and pharmacodynamics of several commonly prescribed medications. The list is not exhaustive nor could it be, since new interactions are continuously being reported. The drugs listed in this table are based on either drug interaction case reports or studies, or potential interactions due to the expected magnitude and seriousness of the interaction (i.e., those identified as contraindicated). Please note that drugs may be listed under specific name, family or pharmacologic class. Reading the entire section is recommended.
Risk of liver damage
The concomitant use of salicylates should be avoided in children under 3 years of age due to the risk of liver toxicity (see 3 SERIOUS WARNINGS AND PRECAUTIONS BOX, Hepatotoxicity, 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Serious or Fatal Hepatotoxicity, and 8.1 Adverse Reaction Overview, Pediatric Population).
Concomitant use of valproate and multiple anticonvulsant therapy increases the risk of liver damage, especially in young children (see 3 SERIOUS WARNINGS AND PRECAUTIONS BOX, Hepatotoxicity, 7 WARNINGS AND PRECAUTIONS, Hepatic/Biliary/Pancreatic, Serious or Fatal Hepatotoxicity, and 8.1 Adverse Reaction Overview, Pediatric Population).
Table 3. Established or Potential Drug-Drug Interactions:
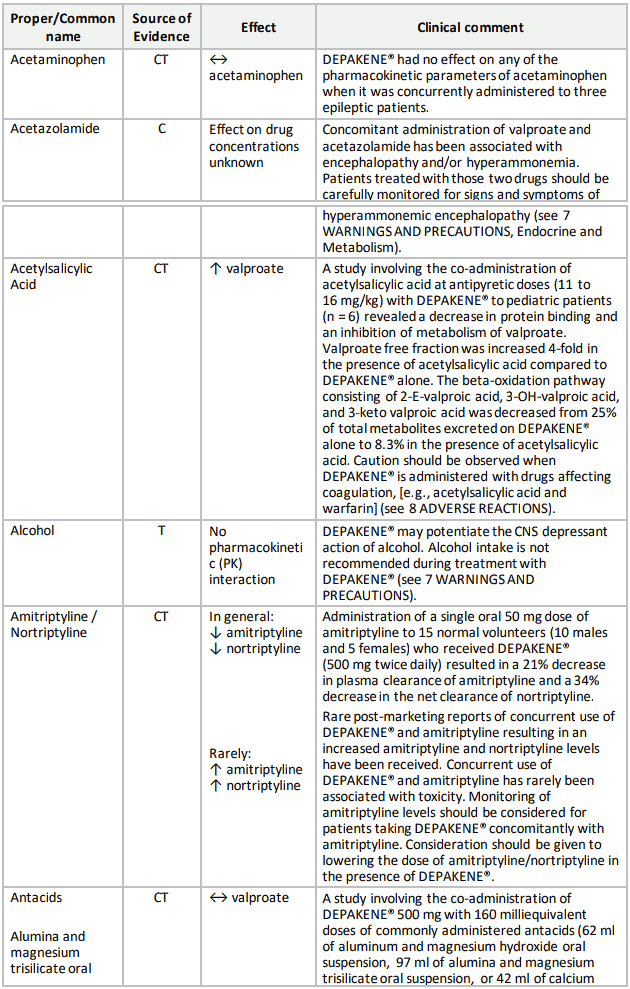
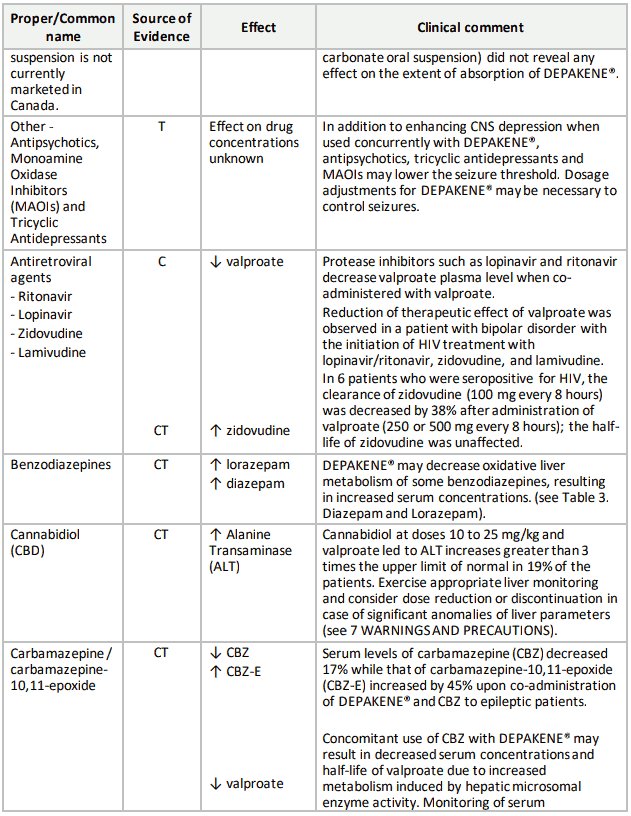
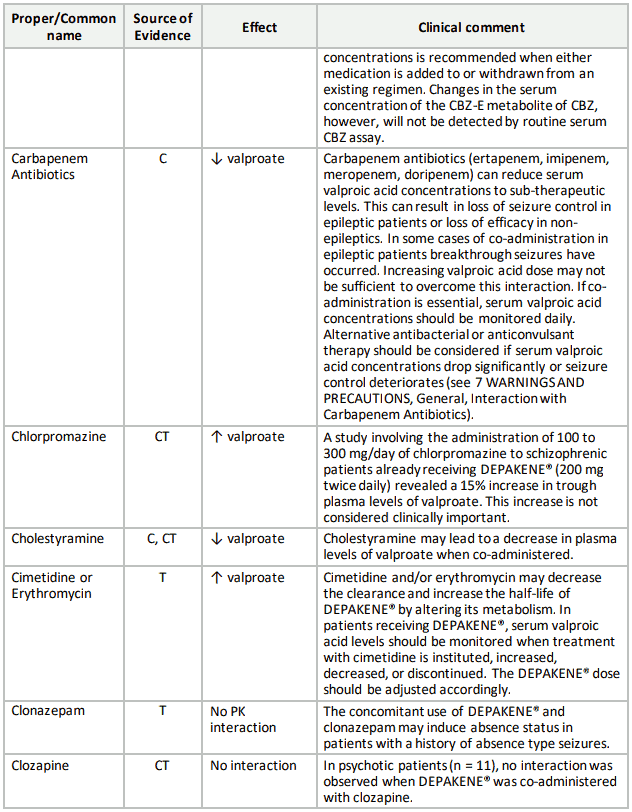
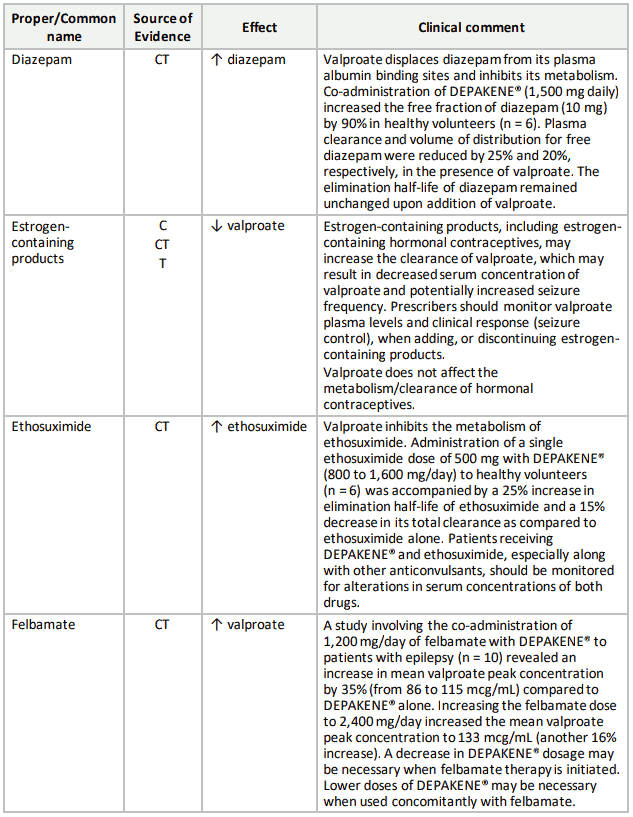

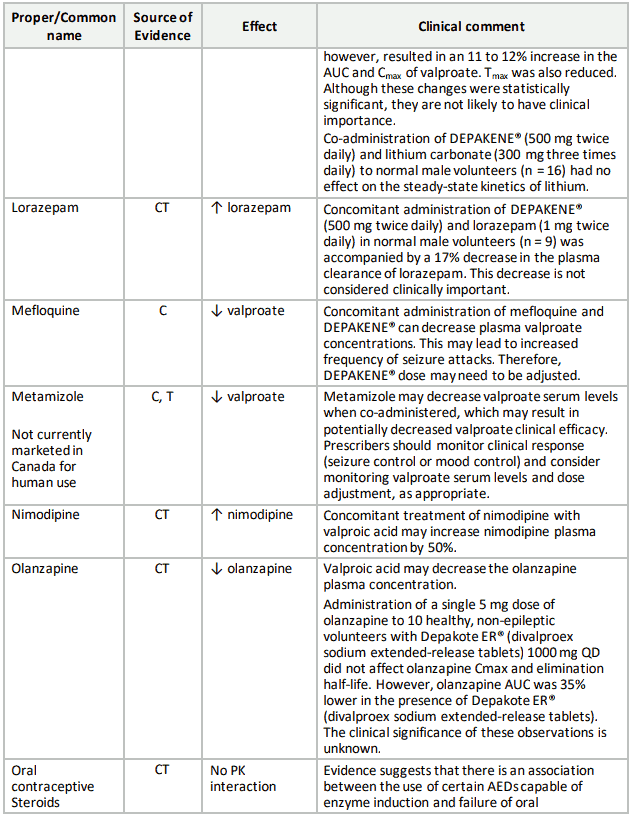
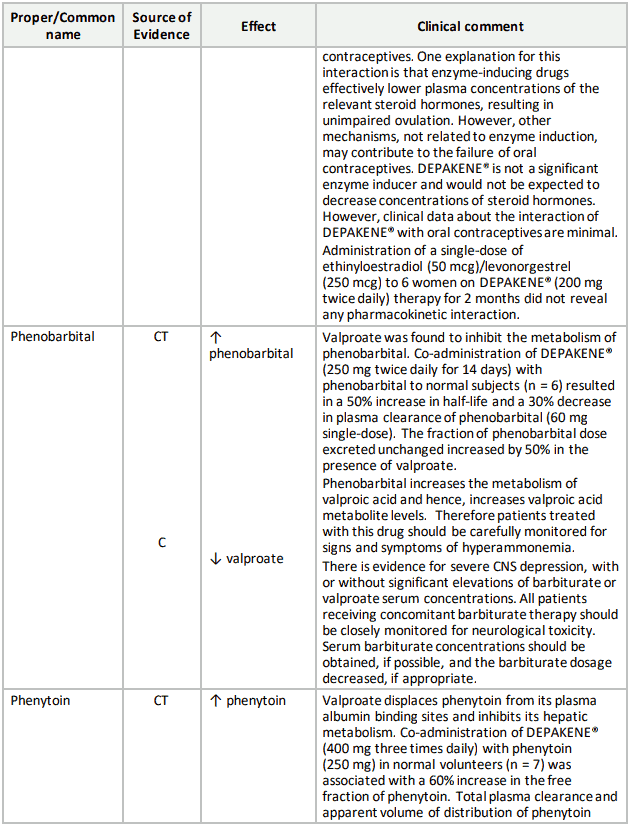
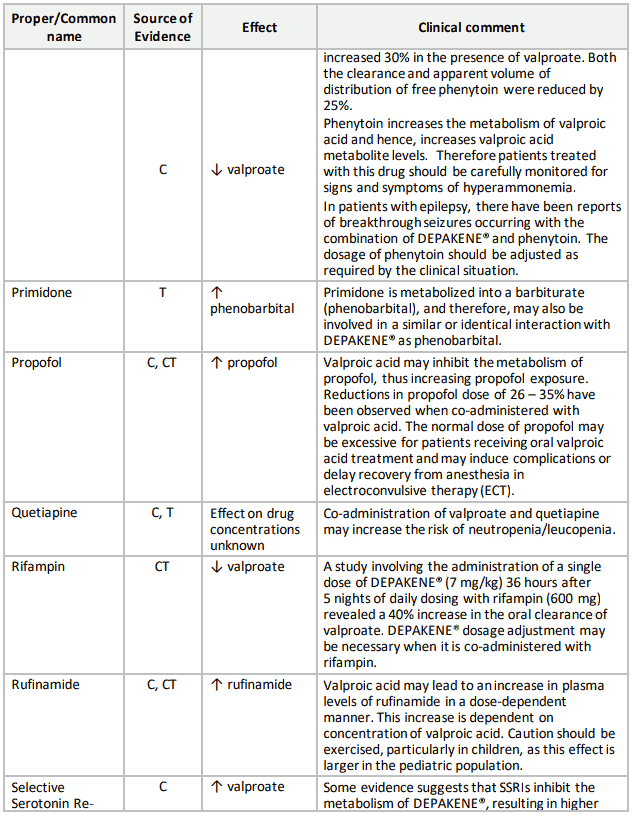
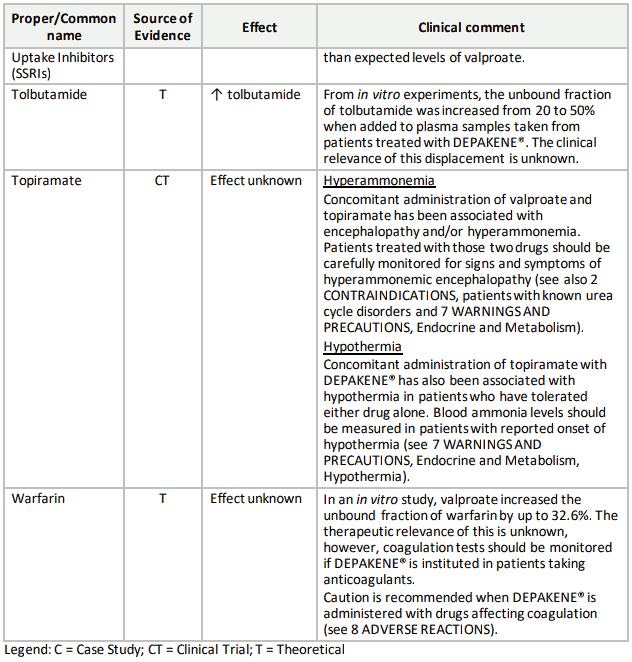
9.5 Drug-Food Interactions
Co-administration of DEPAKENE with food should cause no clinical problems in the management of patients with epilepsy (see 4.4 Administration).
9.6 Drug-Herb Interactions
Interactions with herbal products have not been established.
9.7 Drug-Laboratory Test Interactions
DEPAKENE is partially eliminated in the urine as a ketone-containing metabolite which may lead to a false interpretation of the urine ketone test.
There have been reports of altered thyroid function tests associated with DEPAKENE; the clinical significance of these is unknown (see 7.1.1 Pregnant Women, Thyroid Gland Abnormalities).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.