DEPAKOTE Delayed-release tablet Ref.[10553] Active ingredients: Valproic acid
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Divalproex sodium dissociates to the valproate ion in the gastrointestinal tract. The mechanisms by which valproate exerts its therapeutic effects have not been established. It has been suggested that its activity in epilepsy is related to increased brain concentrations of gamma-aminobutyric acid (GABA).
12.2. Pharmacodynamics
The relationship between plasma concentration and clinical response is not well documented. One contributing factor is the nonlinear, concentration dependent protein binding of valproate which affects the clearance of the drug. Thus, monitoring of total serum valproate cannot provide a reliable index of the bioactive valproate species.
For example, because the plasma protein binding of valproate is concentration dependent, the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Higher than expected free fractions occur in the elderly, in hyperlipidemic patients, and in patients with hepatic and renal diseases.
Epilepsy
The therapeutic range in epilepsy is commonly considered to be 50 to 100 mcg/mL of total valproate, although some patients may be controlled with lower or higher plasma concentrations.
Mania
In placebo-controlled clinical trials of acute mania, patients were dosed to clinical response with trough plasma concentrations between 50 and 125 mcg/mL [see Dosage and Administration (2.1)].
12.3. Pharmacokinetics
Absorption/Bioavailability
Equivalent oral doses of Depakote (divalproex sodium) products and Depakene (valproic acid) capsules deliver equivalent quantities of valproate ion systemically. Although the rate of valproate ion absorption may vary with the formulation administered (liquid, solid, or sprinkle), conditions of use (e.g., fasting or postprandial) and the method of administration (e.g., whether the contents of the capsule are sprinkled on food or the capsule is taken intact), these differences should be of minor clinical importance under the steady state conditions achieved in chronic use in the treatment of epilepsy.
However, it is possible that differences among the various valproate products in Tmax and Cmax could be important upon initiation of treatment. For example, in single dose studies, the effect of feeding had a greater influence on the rate of absorption of the tablet (increase in Tmax from 4 to 8 hours) than on the absorption of the sprinkle capsules (increase in Tmax from 3.3 to 4.8 hours).
While the absorption rate from the G.I. tract and fluctuation in valproate plasma concentrations vary with dosing regimen and formulation, the efficacy of valproate as an anticonvulsant in chronic use is unlikely to be affected. Experience employing dosing regimens from once-a-day to four-times-a-day, as well as studies in primate epilepsy models involving constant rate infusion, indicate that total daily systemic bioavailability (extent of absorption) is the primary determinant of seizure control and that differences in the ratios of plasma peak to trough concentrations between valproate formulations are inconsequential from a practical clinical standpoint. Whether or not rate of absorption influences the efficacy of valproate as an antimanic or antimigraine agent is unknown.
Co-administration of oral valproate products with food and substitution among the various Depakote and Depakene formulations should cause no clinical problems in the management of patients with epilepsy [see Dosage and Administration (2.2)]. Nonetheless, any changes in dosage administration, or the addition or discontinuance of concomitant drugs should ordinarily be accompanied by close monitoring of clinical status and valproate plasma concentrations.
Distribution
Protein Binding
The plasma protein binding of valproate is concentration dependent and the free fraction increases from approximately 10% at 40 mcg/mL to 18.5% at 130 mcg/mL. Protein binding of valproate is reduced in the elderly, in patients with chronic hepatic diseases, in patients with renal impairment, and in the presence of other drugs (e.g., aspirin). Conversely, valproate may displace certain protein-bound drugs (e.g., phenytoin, carbamazepine, warfarin, and tolbutamide) [see Drug Interactions (7.2) for more detailed information on the pharmacokinetic interactions of valproate with other drugs].
CNS Distribution
Valproate concentrations in cerebrospinal fluid (CSF) approximate unbound concentrations in plasma (about 10% of total concentration).
Metabolism
Valproate is metabolized almost entirely by the liver. In adult patients on monotherapy, 30-50% of an administered dose appears in urine as a glucuronide conjugate. Mitochondrial β-oxidation is the other major metabolic pathway, typically accounting for over 40% of the dose. Usually, less than 15-20% of the dose is eliminated by other oxidative mechanisms. Less than 3% of an administered dose is excreted unchanged in urine.
The relationship between dose and total valproate concentration is nonlinear; concentration does not increase proportionally with the dose, but rather, increases to a lesser extent due to saturable plasma protein binding. The kinetics of unbound drug are linear.
Elimination
Mean plasma clearance and volume of distribution for total valproate are 0.56 L/hr/1.73 m² and 11 L/1.73 m², respectively. Mean plasma clearance and volume of distribution for free valproate are 4.6 L/hr/1.73 m² and 92 L/1.73 m². Mean terminal half-life for valproate monotherapy ranged from 9 to 16 hours following oral dosing regimens of 250 to 1,000 mg.
The estimates cited apply primarily to patients who are not taking drugs that affect hepatic metabolizing enzyme systems. For example, patients taking enzyme-inducing antiepileptic drugs (carbamazepine, phenytoin, and phenobarbital) will clear valproate more rapidly. Because of these changes in valproate clearance, monitoring of antiepileptic concentrations should be intensified whenever concomitant antiepileptics are introduced or withdrawn.
Special Populations
Effect of Age
Neonates:
Children within the first two months of life have a markedly decreased ability to eliminate valproate compared to older children and adults. This is a result of reduced clearance (perhaps due to delay in development of glucuronosyltransferase and other enzyme systems involved in valproate elimination) as well as increased volume of distribution (in part due to decreased plasma protein binding). For example, in one study, the half-life in children under 10 days ranged from 10 to 67 hours compared to a range of 7 to 13 hours in children greater than 2 months.
Children:
Pediatric patients (i.e., between 3 months and 10 years) have 50% higher clearances expressed on weight (i.e., mL/min/kg) than do adults. Over the age of 10 years, children have pharmacokinetic parameters that approximate those of adults.
Elderly:
The capacity of elderly patients (age range: 68 to 89 years) to eliminate valproate has been shown to be reduced compared to younger adults (age range: 22 to 26 years). Intrinsic clearance is reduced by 39%; the free fraction is increased by 44%. Accordingly, the initial dosage should be reduced in the elderly [see Dosage and Administration (2.4)].
Effect of Sex
There are no differences in the body surface area adjusted unbound clearance between males and females (4.8±0.17 and 4.7±0.07 L/hr per 1.73 m², respectively).
Effect of Race
The effects of race on the kinetics of valproate have not been studied.
Effect of Disease
Liver Disease:
Liver disease impairs the capacity to eliminate valproate. In one study, the clearance of free valproate was decreased by 50% in 7 patients with cirrhosis and by 16% in 4 patients with acute hepatitis, compared with 6 healthy subjects. In that study, the half-life of valproate was increased from 12 to 18 hours. Liver disease is also associated with decreased albumin concentrations and larger unbound fractions (2 to 2.6 fold increase) of valproate. Accordingly, monitoring of total concentrations may be misleading since free concentrations may be substantially elevated in patients with hepatic disease whereas total concentrations may appear to be normal [see Boxed Warning, Contraindications (4), and Warnings and Precautions (5.1)].
Renal Disease:
A slight reduction (27%) in the unbound clearance of valproate has been reported in patients with renal failure (creatinine clearance <10 mL/minute); however, hemodialysis typically reduces valproate concentrations by about 20%. Therefore, no dosage adjustment appears to be necessary in patients with renal failure. Protein binding in these patients is substantially reduced; thus, monitoring total concentrations may be misleading.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Valproate was administered orally to rats and mice at doses of 80 and 170 mg/kg/day (less than the maximum recommended human dose on a mg/m² basis) for two years. The primary findings were an increase in the incidence of subcutaneous fibrosarcomas in high-dose male rats receiving valproate and a dose-related trend for benign pulmonary adenomas in male mice receiving valproate.
Mutagenesis
Valproate was not mutagenic in an in vitro bacterial assay (Ames test), did not produce dominant lethal effects in mice, and did not increase chromosome aberration frequency in an in vivo cytogenetic study in rats. Increased frequencies of sister chromatid exchange (SCE) have been reported in a study of epileptic children taking valproate; this association was not observed in another study conducted in adults.
Impairment of Fertility
In chronic toxicity studies in juvenile and adult rats and dogs, administration of valproate resulted in testicular atrophy and reduced spermatogenesis at oral doses of 400 mg/kg/day or greater in rats (approximately equal to or greater than the maximum recommended human dose (MRHD) on a mg/m² basis) and 150 mg/kg/day or greater in dogs (approximately equal to or greater than the MRHD on a mg/m² basis). Fertility studies in rats have shown no effect on fertility at oral doses of valproate up to 350 mg/kg/day (approximately equal to the MRHD on a mg/m² basis) for 60 days.
14. Clinical Studies
14.1 Mania
The effectiveness of Depakote for the treatment of acute mania was demonstrated in two 3-week, placebo controlled, parallel group studies.
(1) Study 1: The first study enrolled adult patients who met DSM-III-R criteria for bipolar disorder and who were hospitalized for acute mania. In addition, they had a history of failing to respond to or not tolerating previous lithium carbonate treatment. Depakote was initiated at a dose of 250 mg tid and adjusted to achieve serum valproate concentrations in a range of 50-100 mcg/mL by day 7. Mean Depakote doses for completers in this study were 1,118, 1,525, and 2,402 mg/day at Days 7, 14, and 21, respectively. Patients were assessed on the Young Mania Rating Scale (YMRS; score ranges from 0-60), an augmented Brief Psychiatric Rating Scale (BPRS-A), and the Global Assessment Scale (GAS). Baseline scores and change from baseline in the Week 3 endpoint (last-observation-carry-forward) analysis were as follows:
Table 6. Study 1:
| YMRS Total Score | |||
|---|---|---|---|
| Group | Baseline1 | BL to Wk 32 | Difference3 |
| Placebo | 28.8 | + 0.2 | |
| Depakote | 28.5 | - 9.5 | 9.7 |
| BPRS-A Total Score | |||
| Group | Baseline1 | BL to Wk 32 | Difference3 |
| Placebo | 76.2 | + 1.8 | |
| Depakote | 76.4 | -17.0 | 18.8 |
| GAS Score | |||
| Group | Baseline1 | BL to Wk 32 | Difference3 |
| Placebo | 31.8 | 0.0 | |
| Depakote | 30.3 | + 18.1 | 18.1 |
1 Mean score at baseline
2 Change from baseline to Week 3 (LOCF)
3 Difference in change from baseline to Week 3 endpoint (LOCF) between Depakote and placebo
Depakote was statistically significantly superior to placebo on all three measures of outcome.
(2) Study 2: The second study enrolled adult patients who met Research Diagnostic Criteria for manic disorder and who were hospitalized for acute mania. Depakote was initiated at a dose of 250 mg tid and adjusted within a dose range of 750-2,500 mg/day to achieve serum valproate concentrations in a range of 40-150 mcg/mL. Mean Depakote doses for completers in this study were 1,116, 1,683, and 2,006 mg/day at Days 7, 14, and 21, respectively. Study 2 also included a lithium group for which lithium doses for completers were 1,312, 1,869, and 1,984 mg/day at Days 7, 14, and 21, respectively. Patients were assessed on the Manic Rating Scale (MRS; score ranges from 11-63), and the primary outcome measures were the total MRS score, and scores for two subscales of the MRS, i.e., the Manic Syndrome Scale (MSS) and the Behavior and Ideation Scale (BIS). Baseline scores and change from baseline in the Week 3 endpoint (last-observation-carry-forward) analysis were as follows:
Table 7. Study 2:
| MRS Total Score | |||
|---|---|---|---|
| Group | Baseline1 | BL to Day 212 | Difference3 |
| Placebo | 38.9 | -4.4 | |
| Lithium | 37.9 | -10.5 | 6.1 |
| Depakote | 38.1 | -9.5 | 5.1 |
| MSS Total Score | |||
| Group | Baseline1 | BL to Day 212 | Difference3 |
| Placebo | 18.9 | -2.5 | |
| Lithium | 18.5 | -6.2 | 3.7 |
| Depakote | 18.9 | -6.0 | 3.5 |
| BIS Total Score | |||
| Group | Baseline1 | BL to Day 212 | Difference3 |
| Placebo | 16.4 | -1.4 | |
| Lithium | 16.0 | -3.8 | 2.4 |
| Depakote | 15.7 | -3.2 | 1.8 |
1 Mean score at baseline
2 Change from baseline to Day 21 (LOCF)
3 Difference in change from baseline to Day 21 endpoint (LOCF) between Depakote and placebo and lithium and placebo
Depakote was statistically significantly superior to placebo on all three measures of outcome. An exploratory analysis for age and gender effects on outcome did not suggest any differential responsiveness on the basis of age or gender.
A comparison of the percentage of patients showing ≥30% reduction in the symptom score from baseline in each treatment group, separated by study, is shown in Figure 1.
Figure 1:
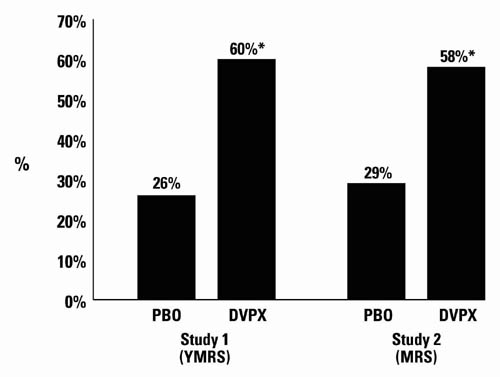
* p<0.05
PBO = placebo, DVPX = Depakote
14.2 Epilepsy
The efficacy of valproate in reducing the incidence of complex partial seizures (CPS) that occur in isolation or in association with other seizure types was established in two controlled trials.
In one, multi-clinic, placebo controlled study employing an add-on design (adjunctive therapy), 144 patients who continued to suffer eight or more CPS per 8 weeks during an 8 week period of monotherapy with doses of either carbamazepine or phenytoin sufficient to assure plasma concentrations within the "therapeutic range" were randomized to receive, in addition to their original antiepilepsy drug (AED), either Depakote or placebo. Randomized patients were to be followed for a total of 16 weeks. The following table presents the findings.
Table 8. Adjunctive Therapy Study Median Incidence of CPS per 8 Weeks:
| Add-on Treatment | Number of Patients | Baseline Incidence | Experimental Incidence |
|---|---|---|---|
| Depakote | 75 | 16.0 | 8.9* |
| Placebo | 69 | 14.5 | 11.5 |
* Reduction from baseline statistically significantly greater for valproate than placebo at p≤0.05 level.
Figure 2 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the adjunctive therapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for an effective treatment is shifted to the left of the curve for placebo. This figure shows that the proportion of patients achieving any particular level of improvement was consistently higher for valproate than for placebo. For example, 45% of patients treated with valproate had a ≥ 50% reduction in complex partial seizure rate compared to 23% of patients treated with placebo.
Figure 2:
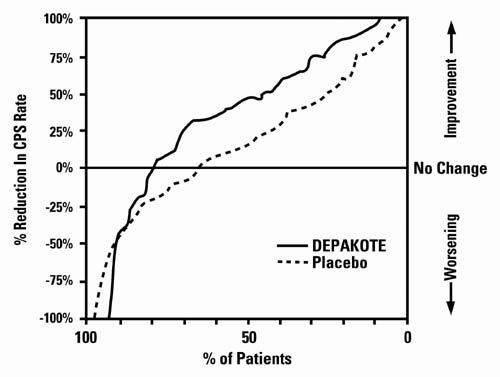
The second study assessed the capacity of valproate to reduce the incidence of CPS when administered as the sole AED. The study compared the incidence of CPS among patients randomized to either a high or low dose treatment arm. Patients qualified for entry into the randomized comparison phase of this study only if 1) they continued to experience 2 or more CPS per 4 weeks during an 8 to 12 week long period of monotherapy with adequate doses of an AED (i.e., phenytoin, carbamazepine, phenobarbital, or primidone) and 2) they made a successful transition over a two week interval to valproate. Patients entering the randomized phase were then brought to their assigned target dose, gradually tapered off their concomitant AED and followed for an interval as long as 22 weeks. Less than 50% of the patients randomized, however, completed the study. In patients converted to Depakote monotherapy, the mean total valproate concentrations during monotherapy were 71 and 123 mcg/mL in the low dose and high dose groups, respectively.
The following table presents the findings for all patients randomized who had at least one post-randomization assessment.
Table 9. Monotherapy Study Median Incidence of CPS per 8 Weeks:
| Treatment | Number of Patients | Baseline Incidence | Randomized Phase Incidence |
|---|---|---|---|
| High dose Depakote | 131 | 13.2 | 10.7* |
| Low dose Depakote | 134 | 14.2 | 13.8 |
* Reduction from baseline statistically significantly greater for high dose than low dose at p≤0.05 level.
Figure 3 presents the proportion of patients (X axis) whose percentage reduction from baseline in complex partial seizure rates was at least as great as that indicated on the Y axis in the monotherapy study. A positive percent reduction indicates an improvement (i.e., a decrease in seizure frequency), while a negative percent reduction indicates worsening. Thus, in a display of this type, the curve for a more effective treatment is shifted to the left of the curve for a less effective treatment. This figure shows that the proportion of patients achieving any particular level of reduction was consistently higher for high dose valproate than for low dose valproate. For example, when switching from carbamazepine, phenytoin, phenobarbital or primidone monotherapy to high dose valproate monotherapy, 63% of patients experienced no change or a reduction in complex partial seizure rates compared to 54% of patients receiving low dose valproate.
Figure 3:
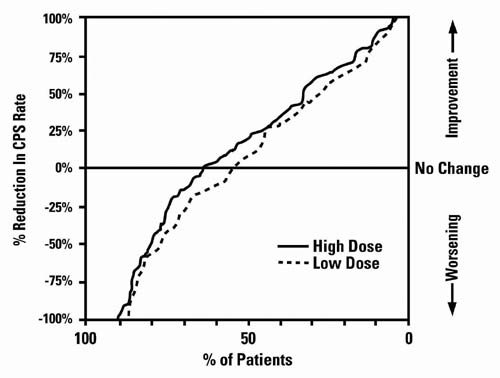
Information on pediatric studies is presented in section 8.
14.3 Migraine
The results of two multicenter, randomized, double-blind, placebo-controlled clinical trials established the effectiveness of Depakote in the prophylactic treatment of migraine headache.
Both studies employed essentially identical designs and recruited patients with a history of migraine with or without aura (of at least 6 months in duration) who were experiencing at least 2 migraine headaches a month during the 3 months prior to enrollment. Patients with cluster headaches were excluded. Women of childbearing potential were excluded entirely from one study, but were permitted in the other if they were deemed to be practicing an effective method of contraception.
In each study following a 4-week single-blind placebo baseline period, patients were randomized, under double blind conditions, to Depakote or placebo for a 12-week treatment phase, comprised of a 4-week dose titration period followed by an 8-week maintenance period. Treatment outcome was assessed on the basis of 4-week migraine headache rates during the treatment phase.
In the first study, a total of 107 patients (24 M, 83 F), ranging in age from 26 to 73 were randomized 2:1, Depakote to placebo. Ninety patients completed the 8-week maintenance period. Drug dose titration, using 250 mg tablets, was individualized at the investigator's discretion. Adjustments were guided by actual/sham trough total serum valproate levels in order to maintain the study blind. In patients on Depakote doses ranged from 500 to 2,500 mg a day. Doses over 500 mg were given in three divided doses (TID). The mean dose during the treatment phase was 1,087 mg/day resulting in a mean trough total valproate level of 72.5 mcg/mL, with a range of 31 to 133 mcg/mL.
The mean 4-week migraine headache rate during the treatment phase was 5.7 in the placebo group compared to 3.5 in the Depakote group (see Figure 4). These rates were significantly different.
In the second study, a total of 176 patients (19 males and 157 females), ranging in age from 17 to 76 years, were randomized equally to one of three Depakote dose groups (500, 1,000, or 1,500 mg/day) or placebo. The treatments were given in two divided doses (BID). One hundred thirty-seven patients completed the 8-week maintenance period. Efficacy was to be determined by a comparison of the 4-week migraine headache rate in the combined 1,000/1,500 mg/day group and placebo group.
The initial dose was 250 mg daily. The regimen was advanced by 250 mg every 4 days (8 days for 500 mg/day group), until the randomized dose was achieved. The mean trough total valproate levels during the treatment phase were 39.6, 62.5, and 72.5 mcg/mL in the Depakote 500, 1,000, and 1,500 mg/day groups, respectively.
The mean 4-week migraine headache rates during the treatment phase, adjusted for differences in baseline rates, were 4.5 in the placebo group, compared to 3.3, 3.0, and 3.3 in the Depakote 500, 1,000, and 1,500 mg/day groups, respectively, based on intent-to-treat results (see Figure 4). Migraine headache rates in the combined Depakote 1,000/1,500 mg group were significantly lower than in the placebo group.
Figure 4. Mean 4-week Migraine Rates:
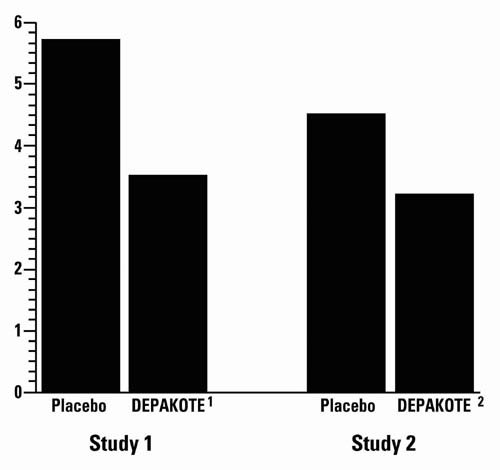
1 Mean dose of Depakote was 1,087 mg/day.
2 Dose of Depakote was 500 or 1,000 mg/day.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.