DETRUNORM Film-coated tablet Ref.[9724] Active ingredients: Propiverine
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2018 Publisher: Amdipharm UK Ltd., Capital House, 85 King William Street, London, EC4N 7BL, United Kingdom
Pharmacodynamic properties
Pharmacotherapeutic group: Urologicals, drugs for urinary frequency and incontinence
ATC code: G04BD06
Mechanism of action
Inhibition of calcium influx and modulation of intracellular calcium in urinary bladder smooth muscle cells causing musculotropic spasmolysis.
Inhibition of the efferent connection of the nervus pelvicus due to anticholinergic action.
Pharmacodynamic effects
In animal models propiverine hydrochloride causes a dose-dependent decrease of the intravesical pressure and an increase in bladder capacity.
The effect is based on the sum of the pharmacological properties of propiverine and three active urinary metabolites as shown in isolated detrusor strips of human and animal origin.
Pharmacokinetic properties
General characteristics of the active substance
Propiverine is nearly completely absorbed from the gastrointestinal tract. It undergoes extensive first pass metabolism. Effects on urinary bladder smooth muscle cells are due to the parent compound and three active metabolites as well, which are rapidly excreted into the urine.
Absorption
Bioequivalence of Detrunorm 15 mg film-coated tablets with the reference medicinal product Mictonorm has been demonstrated by an appropriate bioavailability study.
After oral administration of Mictonorm, propiverine is rapidly absorbed from the gastrointestinal tract with maximal plasma concentrations reached after 2.3 hours. The mean absolute bioavailability of Mictonorm is 40.5% (arithmetic mean value for AUC0-∞ (p.o.) / AUC0-∞ (i.v.)).
Food intake increases the bioavailability of propiverine (mean increase 1.3fold), but does not significantly affect the maximum plasma concentrations of propiverine or of its main metabolite, propiverine-N-oxide. This difference in bioavailability is unlikely to be of clinical significance but adjustment of dose in relation to food intake could be required in patients suffering from impaired renal or hepatic function. Therefore, a regular intake before meals is recommended.
Distribution
After administration of Mictonorm t.i.d., steady state is reached after four to five days at a higher concentration level than after single dose application (Caverage = 61 ng/ml). The volume of distribution was estimated in 21 healthy volunteers after intravenous administration of propiverine hydrochloride to range from 125 to 473 l (mean 279 l) indicating, that a large amount of available propiverine is distributed to peripheral compartments. The binding to plasma proteins is 90-95% for the parent compound and about 60% for the main metabolite.
Plasma concentrations of propiverine in 16 healthy volunteers after single and repeated administration of Mictonorm (t.i.d. for 6 days):
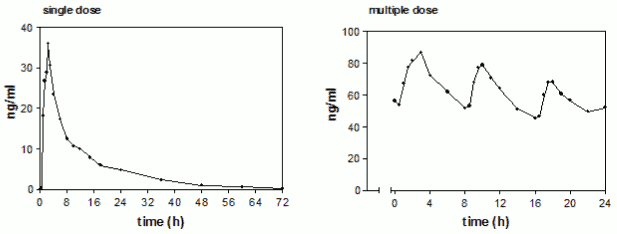
Steady state characteristics of propiverine following multiple-dose administration to 16 healthy volunteers of Mictonorm (t.i.d. for 6 days):
| Dose interval | AUC0-τ | PTF | Caverage | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [h] | [n·gh/ml] | CV [%] | %] | CV [%] | /ml] | CV [%] | 0-8 | 515 | 35 | 57 | 16 | 64 | 36 |
| 8-16 | 460 | 33 | 70 | 25 | 57 | 33 | |||||||
| 16-24 | 421 | 36 | 52 | 39 | 52 | 36 | |||||||
CV: coefficient of variation
PTF: peak-trough fluctuation
Biotransformation
Propiverine is extensively metabolised by intestinal and hepatic enzymes. The primary metabolic route involves the oxidation of the piperidyl-N and is mediated by CYP 3A4 and flavin-containing monoxygenases (FMO) 1 and 3 and leads to the formation of the much less active N-oxide, the plasma concentration of which greatly exceeds that of the parent substance. Four metabolites were identified in urine; three of them are pharmacologically active and may contribute to the therapeutic efficacy of Mictonorm.
In vitro there is a slight inhibition of CYP 3A4 and CYP 2D6 detectable which occurs at concentrations exceeding therapeutic plasma concentrations 10- to 100-fold (see section 4.5).
Elimination
Following administration of 30 mg oral dose of 14C-propiverine hydrochloride to healthy volunteers, 60% of radioactivity was recovered in urine and 21% was recovered in faeces within 12 days. Less than 1% of an oral dose is excreted unchanged in the urine. Mean total clearance after single dose administration of 30 mg is 371 ml/min (191–870 ml/min). In three studies including a total of 37 healthy volunteers the mean elimination half-life was 14.1, 20.1, and 22.1 hours, respectively.
Linearity/ non-linearity
Pharmacokinetic parameters of propiverine and propiverine-N-oxide following oral administration of 10-30 mg of propiverine hydrochloride are linearly related to dose. There are no changes of pharmacokinetics during steady state compared to single dose administration.
Characteristics in patients
Renal impairment
Severe renal impairment does not significantly alter the disposition of propiverine and its main metabolite, propiverine-N-oxide, as deduced from a single dose study in 12 patients with creatinine clearance <30 ml/min. No dose adjustment is to be recommended as long as the total daily dose does not exceed 30 mg (i.e. Detrunorm 15 mg film-coated tablets given b.i.d.). In case that higher dose (i.e. 45 mg) shall be administered a careful titration of dose is recommended considering anticholinergic effects as a marker for tolerability.
Hepatic insufficiency
There were similar steady state pharmacokinetics in 12 patients with mild to moderate impairment of liver function due to fatty liver disease as compared to 12 healthy controls. No data are available for severe hepatic impairment.
Age
The comparison of trough plasma concentrations during steady state (Mictonorm t.i.d. for 28 days) reveals no difference between older patients (60–85 years; mean 68) and young healthy subjects. The ratio of parent drug to metabolite remains unchanged in older patients indicating the metabolic conversion of propiverine to its main metabolite, propiverine-N-oxide, not to be an age-related or limiting step in the overall excretion.
Patients with glaucoma
Intraocular pressure in patients with open angle glaucoma and in patients with treated (controlled) angle closure glaucoma is not increased by Mictonorm t.i.d., as demonstrated by two placebo-controlled studies.
Preclinical safety data
In long term oral dose studies in two mammalian species the main treatment related effect were changes in the liver (including elevation of hepatic enzymes). These were characterised by hepatic hypertrophy and fatty degeneration. The fatty degeneration was reversible upon cessation of treatment.
No effects on male and female fertility and reproduction behaviour were observed in toxicological studies with rats.
In animal studies, skeletal retardation in the offspring occurred when the drug was administered orally at high doses to pregnant females.
In lactating mammals propiverine was excreted into the milk.
There was no evidence of mutagenicity. The carcinogenicity study in mice demonstrated an increased incidence of hepatocellular adenoma and carcinoma in high dose males. In the rat carcinogenicity study hepatocellular adenoma, kidney adenoma and urinary bladder papilloma has been demonstrated in high dose male rats, while in female animals endometrial stromal polyps were increased at the high dose levels. Both the rat and mouse tumours were considered to be species specific and therefore not of clinical relevance.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.