ELVANSE Capsule, hard Ref.[7642] Active ingredients: Lisdexamfetamine
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2019 Publisher: Shire Pharmaceutical Contracts Limited, 1 Kingdom Street, London, W2 6BD, UNITED KINGDOM
Pharmacodynamic properties
Pharmacotherapeutic group: Centrally Acting Sympathomimetics
ATC code: N06BA12
Mechanism of action
Elvanse is a pharmacologically inactive prodrug. After oral administration, lisdexamfetamine is rapidly absorbed from the gastrointestinal tract and hydrolysed primarily by red blood cells to dexamfetamine, which is responsible for the drug's activity.
Amfetamines are non-catecholamine sympathomimetic amines with CNS stimulant activity. The mode of therapeutic action of amfetamine in ADHD is not fully established, however it is thought to be due to its ability to block the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space. The prodrug, lisdexamfetamine, does not bind to the sites responsible for the reuptake of norepinephrine and dopamine in vitro.
Clinical efficacy and safety
The effects of Elvanse in the treatment of ADHD has been demonstrated in three controlled trials in children aged 6 to 12 years, three controlled studies in adolescents aged 13 to 17 years, three controlled studies in children and adolescents (6 to 17 years), and four controlled trials in adults who met the DSM-IV-TR criteria for ADHD.
In clinical studies conducted in children and adults, the effects of Elvanse were ongoing at 13 hours after dosing in children and at 14 hours in adults when the product was taken once daily in the morning.
Paediatric population
Three hundred and thirty-six patients aged 6-17 years were evaluated in the pivotal Phase 3 European Study SPD489-325. In this seven-week randomised double-blind, dose-optimised, placebo- and active-controlled study, Elvanse showed significantly greater efficacy than placebo.
The ADHD Rating Scale is a measure of the core symptoms of ADHD. The placebo-adjusted mean reduction from baseline in patients treated with Elvanse on the ADHD-RS-IV Total Score was 18.6 (p<0.001). At every on-treatment visit and at Endpoint the percentages of subjects who met pre-defined response criteria (a ≥30% reduction from Baseline in ADHD-RS-IV Total Score and a CGI-I value of 1 or 2) was significantly higher (p<0.001) for Elvanse when compared to placebo. The endpoint of this study is defined in Table 1. The results were also significantly higher for Elvanse when compared to placebo when the individual components of the response criteria were evaluated. In addition, mean scores for ADHD symptoms following treatment discontinuation did not exceed baseline scores prior to treatment, indicating there was no rebound effect.
In addition to a reduction in symptoms, clinical studies have demonstrated that Elvanse significantly improves functional outcomes. Specifically, in Study SPD489-325, 75.0% of subjects on Elvanse showed Improvement (defined as "very much improved" or "much improved") on the Clinical Global Impression-Improvement (CGI-I) rating scale compared to 14.2% on placebo (p<0.001).
Elvanse showed significant improvement in child achievement in academic performance, as measured by the Health Related Quality of life instrument, Parent Report Form of the Child Health and Illness Profile-Child Edition (CHIP-CE:PRF) Achievement Domain. Elvanse demonstrated a significant improvement from baseline compared to placebo (Elvanse: 9.4 versus Placebo -1.1) with a mean difference between the two treatment groups of 10.5 (p<0.001).
Table 1. Outcome Results for Study SPD489-325 at Endpoint1 (Full Analysis Set):
| Lisdexamfetamine dimesylate | Placebo | Methylphenidate hydrochloride | |
|---|---|---|---|
| Change in ADHD-RS IV Total Score | |||
| Least Square Mean | -24.3 | -5.7 | -18.7 |
| Effect size (versus Placebo) | 1.804 | N/A | 1.263 |
| P-value (versus Placebo) | <0.001 | N/A | <0.001 |
| ADHD-RS-IV Responders | |||
| Patients Showing a response2 | 83.7% (87/104) | 22.6% (24/106) | 68.2% (73/107) |
| Difference in response from placebo | 61.0 | N/A | 45.6 |
| P-value (versus Placebo) | <0.001 | N/A | <0.001 |
| CGI-I Responders | |||
| Patients Showing Improvement3 | 75.0% (78/104) | 14.2% (15/106) | 58.9% (63/107) |
| Difference in improvement from placebo | 60.8 | N/A | 44.7 |
| P-value (versus Placebo) | <0.001 | N/A | <0.001 |
| Change in CHIP-CE: PRF Achievement Domain | |||
| Least Square Mean | 9.4 | -1.1 | 6.4 |
| Effect size (versus Placebo) | 1.280 | N/A | 0.912 |
| P-value (versus Placebo) | <0.001 | N/A | <0.001 |
1 Endpoint = the last on-treatment post-Baseline visit of the dose optimisation or dose maintenance Period (Visits 1-7) with a valid value
2 Response is defined as percentage reduction from Baseline in the ADHD-RS-IV Total Score of ≥30%
3 Improvement ("very much improved" or "much improved")
A double-blind, randomised, active-controlled, dose-optimisation study was conducted in children and adolescents aged 6 to 17 years (n=267) who met DSM-IV criteria for ADHD. In this nine-week study, patients were randomised (1:1) to a daily morning dose of Elvanse (30, 50 or 70 mg/day), or atomoxetine (dosed as appropriate for the subject's weight up to 100 mg). During a 4-week Dose Optimisation Period, patients were titrated until an optimal dose, based on treatment emergent adverse events and clinical judgement, was reached. Patients treated with Elvanse had a shorter time to first response compared to patients treated with atomoxetine (median 13.0 vs 21.0 days, respectively; p=0.003), where response was defined as having a CGI-I score of 1 (very much improved) or 2 (much improved) at any of the double-blind treatment visits. Across all of the double blind treatment visits, the proportion of responders in the Elvanse group was consistently higher than the proportion of responders in the atomoxetine group. The difference ranged from 16-24 percentage points. At the study endpoint the least square mean changes from baseline in ADHD-RS-IV Total Score for Elvanse and atomoxetine were -26.1 and -19.7, respectively, with a between-group difference of -6.4.
Two double-blind, parallel-group, active-controlled (OROS-MPH [Concerta]) studies have been conducted in adolescents aged 13-17 years with ADHD. Both studies also included a placebo reference arm. The 8-week dose-optimization study (SPD489-405) had a 5-week dose-optimization period and a 3-week dose-maintenance period. During the dose-optimization period, subjects were titrated once weekly based on TEAEs and clinical response to an optimal dose of 30, 50, or 70 mg/day (for SPD489 subjects) or 18, 36, 54, or 72 mg/day (for OROS-MPH subjects), which was maintained throughout a 3-week dose-maintenance period. The mean doses at endpoint were 57.9 mg and 55.8 mg for SPD489 and OROS-MPH, respectively. In this study, neither SPD489 nor OROS-MPH was found to be statistically superior to the other product at Week 8. The 6-week fixed-dose study (SPD489-406) had a 4-week forced-dose titration period and a 2-week dose-maintenance period. At the highest doses of SPD489 (70 mg) and OROS-MPH (72 mg), SPD489 treatment was found to be superior to OROS-MPH as measured by both the primary efficacy analysis (change from baseline at Week 6 on the ADHD-RS Total score) and the key secondary efficacy analysis (at last study visit on the CGI-I) (see Table 2).
Table 2. Change from Baseline on ADHD-RS-IV Total Score and Endpoint on CGI-I (Full Analysis Set):
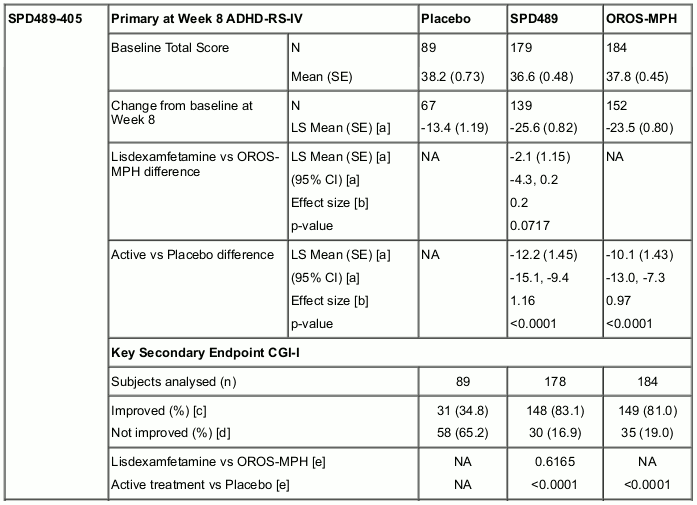
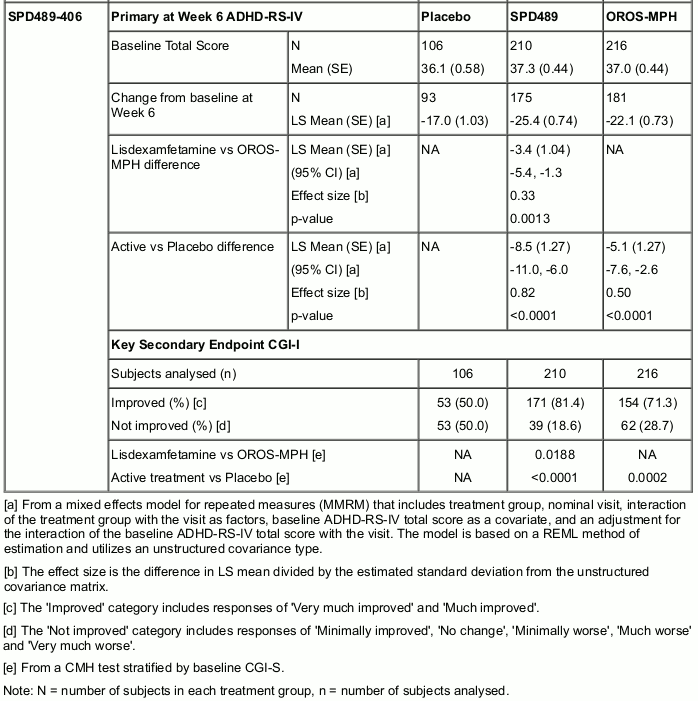
A 2-year open label safety study conducted in children and adolescents (ages 6-17) with ADHD enrolled 314 patients. Of these, 191 patients completed the study.
In addition, maintenance of effect was demonstrated in a double-blind, placebo-controlled, randomised withdrawal study conducted in children and adolescents ages 6 to 17 (n=157) who met the diagnosis of ADHD (DSM-IV criteria). Patients were optimised to open-label Elvanse for an extended period (at least 26 weeks) prior to entry into the 6-week randomised withdrawal period. Eligible patients were randomised to continue receiving their optimised dose of Elvanse or to switch to placebo. Patients were observed for relapse (treatment failure) during the 6-week double-blind phase. Treatment failure was defined as a ≥50% increase (worsening) in the ADHD-RS Total Score and a ≥2-point increase in the CGI-S score compared to scores at entry into the double-blind randomised withdrawal phase. Treatment failure was significantly lower (p<0.001) for the Elvanse subjects (15.8%) compared to placebo (67.5%). For the majority of subjects (70.3%) who were treatment failures regardless of treatment, ADHD symptoms worsened at or before the week 2 visit following randomisation.
Adult population
The effectiveness of Elvanse in the treatment of ADHD was established in a double-blind, randomised, placebo-controlled, parallel-group study conducted in 420 adult patients aged 18 to 55 years who met DSM-IV criteria for ADHD. Significant improvements in ADHD symptoms, based upon investigator ratings on the ADHD-RS with adult prompts total score, were observed for all Elvanse doses compared to placebo. Treatment with Elvanse significantly reduced the degree of functional impairment as measured by improvement on the CGI-I rating scale compared to placebo.
In addition, maintenance of effect was demonstrated in a double-blind, placebo-controlled, randomised withdrawal design study that enrolled adults (n=123) who met DSM-IV criteria for ADHD and who, at study entry, had been treated with Elvanse for a minimum of 6 months. A significantly lower proportion of patients treated with Elvanse met relapse criteria (8.9%) compared to patients receiving placebo (75.0%) in the double-blind randomised withdrawal phase. Relapse was defined as a ≥50% increase from randomisation in ADHD-RS-IV Total Score and a ≥2 point increase in CGI-S score relative to the CGI-S score at randomisation.
Abuse liability studies
In a human abuse liability study, when equivalent oral doses of 100 mg lisdexamfetamine dimesylate and 40 mg immediate-release dexamfetamine sulphate were administered to individuals with a history of drug abuse, lisdexamfetamine dimesylate 100 mg produced subjective responses on a scale of "Drug Liking Effects" (primary endpoint) that were significantly less than dexamfetamine immediate-release 40 mg. However, oral administration of 150 mg lisdexamfetamine dimesylate produced increases in positive subjective responses on this scale that were comparable to the positive subjective responses produced by 40 mg of oral immediate-release dexamfetamine and 200 mg of diethylpropion.
Intravenous administration of 50 mg lisdexamfetamine dimesylate to individuals with a history of drug abuse produced positive subjective responses on scales measuring "Drug Liking", "Euphoria", "Amfetamine Effects", and "Benzedrine Effects" that were greater than placebo but less than those produced by an equivalent dose (20 mg) of intravenous dexamfetamine.
Pharmacokinetic properties
Absorption
After oral administration, lisdexamfetamine dimesylate is rapidly absorbed from the gastrointestinal tract of healthy adults and children (6 to 12 years) with ADHD, thought to be mediated by the high capacity PEPT1 transporter.
Food does not affect the observed AUC and Cmax of dexamfetamine in healthy adults after single-dose oral administration of Elvanse 70 mg capsules but prolongs Tmax by approximately 1 hour (from 3.8 hours at fasted state to 4.7 hours after a high fat meal). After an 8-hour fast, the AUCs for dexamfetamine following oral administration of lisdexamfetamine dimesylate in solution and as intact capsules were equivalent.
Distribution
In 18 children (6 to 12 years) with ADHD, the Tmax of dexamfetamine was approximately 3.5 hours following single-dose oral administration of lisdexamfetamine dimesylate either 30 mg, 50 mg, or 70 mg administered after an 8-hour overnight fast. The Tmax of lisdexamfetamine dimesylate was approximately 1 hour. Linear pharmacokinetics of dexamfetamine after single-dose oral administration of lisdexamfetamine dimesylate was established over the dose range of 30 mg to 70 mg in children aged 6 to 12 years.
Weight/dose normalised AUC and Cmax were 22% and 12% lower, respectively, in adult females than in males on day 7 following a 70 mg/day dose of lisdexamfetamine for 7 days. Weight/dose normalised AUC and Cmax values were the same in girls and boys following single doses of 30-70 mg.
There is no accumulation of dexamfetamine at steady state in healthy adults and no accumulation of lisdexamfetamine dimesylate after once-daily dosing for 7 consecutive days.
Biotransformation
Lisdexamfetamine dimesylate is converted to dexamfetamine and l-lysine, which occurs by metabolism in blood primarily due to the hydrolytic activity of red blood cells. Red blood cells have a high capacity for metabolism of lisdexamfetamine as in vitro data demonstrated substantial hydrolysis occurs even at low hematocrit levels. Lisdexamfetamine is not metabolised by cytochrome P450 enzymes.
Amfetamine is oxidised at the 4 position of the benzene ring to form 4-hydroxyamfetamine, or on the side chain α or β carbons to form alpha-hydroxy-amfetamine or norephedrine, respectively. Norephedrine and 4-hydroxy-amfetamine are both active and each is subsequently oxidised to form 4-hydroxy-norephedrine. Alpha-hydroxy-amfetamine undergoes deamination to form phenylacetone, which ultimately forms benzoic acid and its glucuronide and the glycine conjugate hippuric acid. Although the enzymes involved in amfetamine metabolism have not been clearly defined, CYP2D6 is known to be involved with formation of 4-hydroxy-amfetamine.
Elimination
Following the oral administration of a 70 mg dose of radiolabelled lisdexamfetamine dimesylate to 6 healthy subjects, approximately 96% of the oral dose radioactivity was recovered in the urine and only 0.3% recovered in the faeces over a period of 120 hours. Of the radioactivity recovered in the urine 42% of the dose was related to amfetamine, 25% to hippuric acid, and 2% intact lisdexamfetamine. Plasma concentrations of unconverted lisdexamfetamine are low and transient, generally becoming non-quantifiable by 8 hours after administration. The plasma elimination half-life of lisdexamfetamine typically averaged less than one hour in studies of lisdexamfetamine dimesylate in volunteers. The half-life of dexamfetamine is 11 hours.
Special populations
The pharmacokinetics of dexamfetamine, as evaluated by clearance, is similar in children (aged 6 to 12) and adolescents (aged 13 to 17) ADHD patients, and healthy adult volunteers after correcting for body weight.
Systemic exposure to dexamfetamine is similar for men and women given the same mg/kg dose.
Formal pharmacokinetic studies for race have not been conducted. There is no evidence of any impact of ethnicity on the pharmacokinetics of Elvanse.
In a pharmacokinetic study of 40 subjects (8 subjects in each of five renal functional groups: normal, mild impairment, moderate impairment, severe impairment, and end stage renal disease) dexamfetamine clearance was reduced from 0.7 L/hr/kg in normal subjects to 0.4 L/hr/kg in subjects with severe renal impairment (GFR 15 to <30 mL/min1.73m² or CrCl <30 mL/min).
In a study of 47 subjects aged 55 years of age or older amfetamine clearance was approximately 0.7 L/hr/kg for subjects 55 to 74 years of age and 0.55 L/hr/kg for subjects ≥75 years of age. This is slightly reduced compared to younger adults (approximately 1 L/hr/kg for subjects 18 to 45 years of age).
Preclinical safety data
In repeat dose toxicity studies the major findings were changes in behaviour, such as increased activity typical of stimulant administration, with associated reductions in body weight gain, growth measurements and food intake, considered to be a consequence of an exaggerated pharmacological response.
Lisdexamfetamine dimesylate was not genotoxic when tested in vitro in the Ames test and the mouse lymphoma assay or in vivo in the mouse bone marrow micronucleus test. Carcinogenicity studies of lisdexamfetamine dimesylate have not been performed. No evidence of carcinogenicity was found in studies in which d-, l-amfetamine (enantiomer ratio of 1:1) was administered to mice and rats in the diet for 2 years at doses of up to 30 mg/kg/day in male mice, 19 mg/kg/day in female mice, and 5 mg/kg/day in male and female rats.
Lisdexamfetamine dimesylate had no effect on embryofoetal development or survival when administered orally to pregnant rats at doses up to 40 mg/kg/day, and rabbits at doses up to 120 mg/kg/day.
No adverse effects on nervous system development or reproductive function were observed following repeat dose administration of lisdexamfetamine dimesylate to juvenile rats and dogs.
Amfetamine (d- to l-enantiomer ratio of 3:1) did not adversely affect fertility or early embryonic development in the rat at doses of up to 20 mg/kg/day.
A number of studies in rodents indicate that prenatal or early postnatal exposure to amfetamine (d- or d,l-) at doses similar to those used clinically can result in long-term neurochemical and behavioural alterations. Reported behavioural effects include learning and memory deficits, altered locomotor activity, and changes in sexual function. Similar studies have not been conducted for Elvanse.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.