EXVIERA Film-coated tablet Ref.[8766] Active ingredients: Dasabuvir
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: AbbVie Deutschland GmbH & Co. KG, Knollstrasse, 67061 Ludwigshafen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: Antivirals for systemic use; direct-acting antivirals
ATC code: J05AP09
Mechanism of action
Dasabuvir is a non-nucleoside inhibitor of the HCV RNA-dependent RNA polymerase encoded by the NS5B gene, which is essential for replication of the viral genome.
Co-administration of dasabuvir with ombitasvir/paritaprevir/ritonavir combines three direct-acting antiviral medicinal products with distinct mechanisms of action and non-overlapping resistance profiles to target HCV at multiple steps in the viral lifecycle. Refer to the Summary of Product Characteristics of ombitasvir/paritaprevir/ritonavir for its pharmacological properties.
Activity in cell culture and biochemical studies
The EC50 of dasabuvir against genotype 1a-H77 and 1b-Con1 strains in HCV replicon cell culture assays was 7.7 and 1.8 nM, respectively. The replicon activity of dasabuvir was attenuated 12- to 13- fold in the presence of 40% human plasma. The mean EC50 of dasabuvir against replicons containing NS5B from a panel of treatment-naïve genotype 1a and 1b isolates in the HCV replicon cell culture assay was 0.77 nM (range 0.4 to 2.1 nM; n=11) and 0.46 nM (range 0.2 to 2 nM; n=10), respectively. In biochemical assays, dasabuvir inhibited a panel of genotype 1a and 1b polymerases with a mean IC50 value of 4.2 nM (range 2.2 to 10.7 nM; n=7).
The M1 metabolite of dasabuvir had EC50 values of 39 and 8 nM against genotype 1a-H77 and 1bCon1 strains in HCV replicon cell culture assays, respectively, and the activity of the M1 metabolite was attenuated 3- to 4-fold in the presence of 40% human plasma. Dasabuvir had reduced activity in biochemical assays against NS5B polymerases from HCV genotypes 2a, 2b, 3a and 4a (IC50 values ranging from 900 nM to >20 μM).
Resistance
In cell culture
Resistance to dasabuvir conferred by variants in NS5B selected in cell culture or identified in Phase 2b and 3 clinical trials were phenotypically characterised in the appropriate genotype 1a or 1b replicons.
In genotype 1a, substitutions C316Y, M414T, Y448H, A553T, G554S, S556G/R, and Y561H in HCV NS5B reduced susceptibility to dasabuvir. In the genotype 1a replicon, the activity of dasabuvir was reduced 21- to 32-fold by the M414T, S556G or Y561H substitutions; 152- to 261-fold by the A553T, G554S or S556R substitutions; and 1472- and 975-fold by the C316Y and Y448H substitutions, respectively. G558R and D559G/N were observed as treatment-emergent substitutions but the activity of dasabuvir against these variants could not be evaluated due to poor replication capacity. In genotype 1b, substitutions C316N, C316Y, M414T, Y448H, and S556G in HCV NS5B reduced susceptibility to dasabuvir. The activity of dasabuvir was reduced by 5- and 11-fold by C316N and S556G, respectively; 46-fold by M414T or Y448H; and 1569-fold by the C316Y substitutions in the genotype 1b replicon. Dasabuvir retained full activity against replicons containing substitutions S282T in the nucleoside binding site, M423T in the lower thumb site, and P495A/S, P496S or V499A in the upper thumb site.
Effect of baseline HCV substitutions / polymorphisms on treatment response
A pooled analysis of subjects with genotype 1 HCV infection, who were treated with dasabuvir, ombitasvir and paritaprevir with or without ribavirin in Phase 2b and 3 clinical trials, was conducted to explore the association between baseline NS3/4A, NS5A or NS5B substitutions/polymorphisms and treatment outcome in these recommended regimens.
In the greater than 500 genotype 1a baseline samples in this analysis, the most frequently observed resistance-associated variants were M28V (7.4%) in NS5A and S556G (2.9%) in NS5B. Q80K, although a highly prevalent polymorphism in NS3 (41.2% of samples), confers minimal resistance to paritaprevir. Resistance-associated variants at amino acid positions R155 and D168 in NS3 were rarely observed (less than 1%) at baseline. In the greater than 200 genotype 1b baseline samples in this analysis, the most frequently observed resistance-associated variants observed were Y93H (7.5%) in NS5A, and C316N (17.0%) and S556G (15%) in NS5B. Given the low virologic failure rates observed with recommended treatment regimens for HCV genotype 1a- and 1b-infected subjects, the presence of baseline variants appears to have little impact on the likelihood of achieving SVR.
In clinical studies
Of the 2,510 HCV genotype 1 infected subjects who were treated with regimens containing dasabuvir, ombitasvir and paritaprevir with or without ribavirin (for 8, 12 or 24 weeks) in Phase 2b and 3 clinical trials, a total of 74 subjects (3%) experienced virologic failure (primarily post-treatment relapse). Treatment-emergent variants and their prevalence in these virologic failure populations are shown in Table 5. In the 67 genotype 1a infected subjects, NS3 variants were observed in 50 subjects, NS5A variants were observed in 46 subjects, NS5B variants were observed in 37 subjects, and treatmentemergent variants were seen in all 3 drug targets in 30 subjects. In the 7 genotype 1b infected subjects, treatment-emergent variants were observed in NS3 in 4 subjects, in NS5A in 2 subjects, and in both NS3 and NS5A in 1 subject. No genotype 1b infected subjects had treatment-emergent variants in all 3 drug targets.
Table 5. Treatment-emergent amino acid substitutions in the pooled analysis of dasabuvir and ombitasvir/paritaprevir/ritonavir, with and without RBV regimens in Phase 2b and Phase 3 clinical trials (N=2510):
| Target | Emergent amino acid substitutionsa | Genotype 1a N=67b% (n) | Genotype 1b N=7% (n) |
|---|---|---|---|
| NS3 | V55Ic | 6 (4) | - |
| Y56Hc | 9 (6) | 42.9 (3)d | |
| I132Vc | 6 (4) | - | |
| R155K | 13.4 (9) | - | |
| D168A | 6 (4) | - | |
| D168V | 50.7 (34) | 42.9 (3)d | |
| D168Y | 7.5 (5) | - | |
| V36Ac, V36Mc, F43Lc, D168H, E357Kc | <5% | - | |
| NS5A | M28T | 20.9 (14) | - |
| M28Ve | 9 (6) | - | |
| Q30Re | 40.3 (27) | - | |
| Y93H | 28.6 (2) | ||
| H58D, H58P, Y93N | <5% | - | |
| NS5B | A553T | 6.1 (4) | - |
| S556G | 33.3 (22) | - | |
| C316Y, M414T, G554S, S556R, G558R, D559G, D559N, Y561H | <5% | - |
a. Observed in at least 2 subjects of the same subtype.
b. N=66 for the NS5B target.
c. Substitutions were observed in combination with other emergent substitutions at NS3 position R155 or D168.
d. Observed in combination in genotype 1b-infected subjects.
e. Observed in combination in 6% (4/67) of the subjects.
Note: The following variants were selected in cell culture but were not treatment-emergent: NS3 variants A156T in genotype 1a, and R155Q and D168H in genotype 1b; NS5A variants Y93C/H in genotype 1a, and
L31F/V or Y93H in combination with L28M, L31F/V or P58S in genotype 1b; and NS5B variants Y448H in genotype 1a, and M414T and Y448H in genotype 1b.
Persistence of resistance-associated substitutions
The persistence of dasabuvir, ombitasvir and paritaprevir resistance-associated amino acid substitutions in NS5B, NS5A and NS3, respectively, was assessed in genotype 1a-infected subjects in Phase 2b trials. Dasabuvir treatment-emergent variants M414T, G554S, S556G, G558R or D559G/N in NS5B were observed in 34 subjects. Ombitasvir treatment-emergent variants M28T, M28V or Q30R in NS5A were observed in 32 subjects. Paritaprevir treatment-emergent variants V36A/M, R155K or D168V were observed in NS3 in 47 subjects.
NS3 variants V36A/M and R155K and NS5B variants M414T and S556G remained detectable at posttreatment Week 48, whereas NS3 variant D168V and all other NS5B variants were not observed at post-treatment Week 48. All treatment-emergent variants in NS5A remained detectable at posttreatment Week 48. Due to high SVR rates in genotype 1b, trends in persistence of treatment-emergent variants in this genotype could not be established.
The lack of detection of virus containing a resistance-associated substitution does not indicate that the resistant virus is no longer present at clinically significant levels. The long-term clinical impact of the emergence or persistence of virus containing dasabuvir and ombitasvir/paritaprevir/ritonavir - resistance-associated substitutions on future treatment is unknown.
Cross-resistance
Cross-resistance is expected among NS5A inhibitors, NS3/4A protease inhibitors, and non-nucleoside NS5B inhibitors by class. The impact of prior dasabuvir, ombitasvir, or paritaprevir treatment experience on the efficacy of other NS5A inhibitors, NS3/4A protease inhibitors, or NS5B inhibitors has not been studied.
Clinical efficacy and safety
The efficacy and safety of dasabuvir in combination with ombitasvir/paritaprevir/ritonavir with and without ribavirin was evaluated in eight Phase 3 clinical trials, including two trials exclusively in subjects with compensated cirrhosis (Child-Pugh A), in over 2,360 subjects with genotype 1 chronic hepatitis C infection as summarised in Table 6.
Table 6. Phase 3 global multicentre trials conducted with dasabuvir and ombitasvir/paritaprevir/ritonavir with or without ribavirin (RBV):
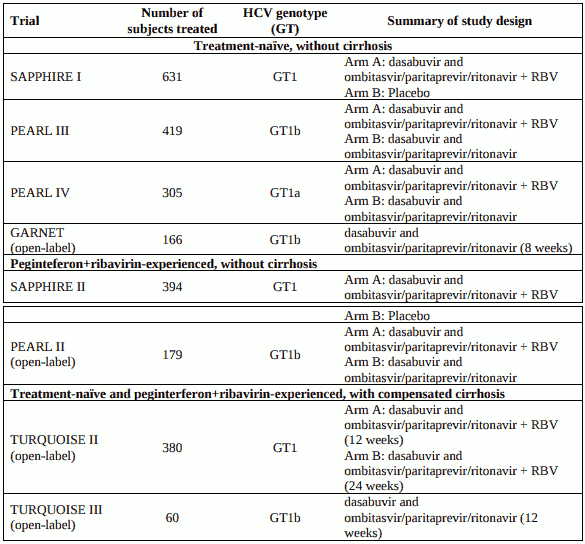
In all eight trials, the dasabuvir dose was 250 mg twice daily and the ombitasvir/paritaprevir/ritonavir dose was 25 mg/150 mg/100 mg once daily. For subjects who received ribavirin, the ribavirin dose was 1000 mg per day for subjects weighing less than 75 kg or 1200 mg per day for subjects weighing greater than or equal to 75 kg.
Sustained virologic response (SVR) was the primary endpoint to determine the HCV cure rate in the Phase 3 studies and was defined as unquantifiable or undetectable HCV RNA 12 weeks after the end of treatment (SVR12). Treatment duration was fixed in each trial and was not guided by subjects' HCV RNA levels (no response guided algorithm). Plasma HCV RNA values were measured during the clinical trials using the COBAS TaqMan HCV test (version 2.0), for use with the High Pure System (except GARNET which used COBAS AmpliPrep/COBAS TaqMan HCV Test v2.0). The High Pure system assay had a lower limit of quantification (LLOQ) of 25 IU per mL and the AmpliPrep assay had a LLOQ of 15 IU per mL.
Clinical trials in treatment-naïve adults
SAPPHIRE-I - genotype 1, treatment-naïve, without cirrhosis
Design: randomised, global multicentre, double-blind, placebo-controlled
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir with weight-based ribavirin for 12 weeks
Treated subjects (N=631) had a median age of 52 years (range: 18 to 70); 54.5% were male; 5.4% were Black; 15.2% had a history of depression or bipolar disorder; 79.1% had baseline HCV RNA levels of at least 800,000 IU/mL; 15.4% had portal fibrosis (F2) and 8.7% had bridging fibrosis (F3); 67.7% had HCV genotype 1a infection; 32.3% had HCV genotype 1b infection.
Table 7. SVR12 for genotype 1-infected treatment-naïve subjects in SAPPHIRE-I:
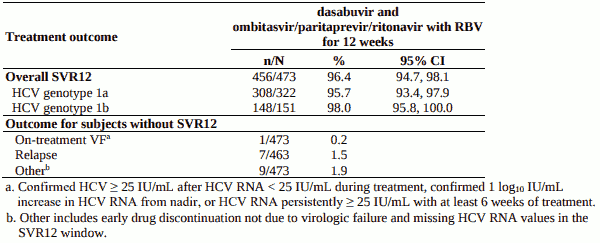
No subjects with HCV genotype 1b infection experienced on-treatment virologic failure and one subject with HCV genotype 1b infection experienced relapse.
PEARL-III - genotype 1b, treatment-naïve, without cirrhosis
Design: randomised, global multicentre, double-blind, regimen-controlled
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir without ribavirin or with weight-based ribavirin for 12 weeks
Treated subjects (N=419) had a median age of 50 years (range: 19 to 70); 45.8% were male; 4.8% were Black; 9.3% had a history of depression or bipolar disorder; 73.3% had baseline HCV RNA of at least 800,000 IU/mL; 20.3% had portal fibrosis (F2) and 10.0% had bridging fibrosis (F3).
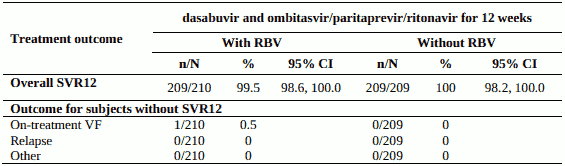
Table 8. SVR12 for genotype 1b-infected treatment-naïve subjects in PEARL III:
PEARL-IV - genotype 1a, treatment-naïve, without cirrhosis
Design: randomised, global multicentre, double-blind, regimen-controlled
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir without ribavirin or with weight-based ribavirin for 12 weeks
Treated subjects (N=305) had a median age of 54 years (range: 19 to 70); 65.2% were male; 11.8% were Black; 20.7% had a history of depression or bipolar disorder; 86.6% had baseline HCV RNA levels of at least 800,000 IU/mL; 18.4% had portal fibrosis (F2) and 17.7% had bridging fibrosis (F3).
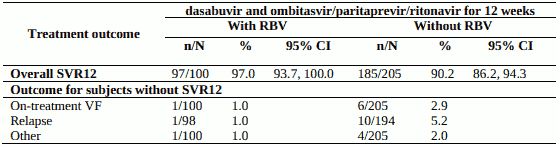
Table 9. SVR12 for genotype 1a-infected treatment-naïve subjects in PEARL IV:
GARNET - Genotype 1b, Treatment-Naïve without cirrhosis.
Design: open-label, single-arm, global multicentre
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir for 8 weeks
Treated subjects (N=166) had a median age of 53 years (range: 22 to 82); 56.6% were female; 3.0% were Asian; 0.6% were Black; 7.2% had baseline HCV RNA levels of at least 6,000,000 IU per mL; 9% had advanced fibrosis (F3) and 98.2% had HCV genotype 1b infection (one subject each had genotype 1a, 1d, and 6 infection).
Table 10. SVR12 for Genotype 1b-infected treatment-naïve subjects without cirrhosis:
| dasabuvir and ombitasvir/paritaprevir/ritonavir for 8 weeks n/N (%) | |
|---|---|
| SVR12 | 160/163 (98.2) |
| 95% CIa | 96.1, 100.0 |
| F0-F1 | 138/139 (99.3)b |
| F2 | 9/9 (100) |
| F3 | 13/15 (86.7)c |
a. Calculated using the normal approximation to the binomial distribution
b. 1 patient discontinued due to non-compliance
c. Relapse in 2/15 patients (confirmed HCV RNA ≥15 IU/mL post-treatment before or during SVR12 window
among subjects with HCV RNA <15 IU/mL at last observation with at least 51 days of treatment).
Clinical trials in peginterferon+ribavirin-experienced adults
SAPPHIRE-II - genotype 1, pegIFN + RBV-experienced, without cirrhosis
Design: randomised, global multicentre, double-blind, placebo-controlled
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir with weight-based ribavirin for 12 weeks
Treated subjects (N=394) had a median age of 54 years (range: 19 to 71); 49.0% were prior pegIFN/RBV null responders; 21.8/% were prior pegIFN/RBV partial responders; and 29.2% were prior pegIFN/RBV relapsers; 57.6% were male; 8.1% were Black; 20.6% had a history of depression or bipolar disorder; 87.1% had baseline HCV RNA levels of at least 800,000 IU per mL; 17.8% had portal fibrosis (F2) and 14.5% had bridging fibrosis (F3); 58.4% had HCV genotype 1a infection; 41.4% had HCV genotype 1b infection.
Table 11. SVR12 for genotype 1-infected peginterferon+ribavirin-experienced subjects in SAPPHIRE-II:
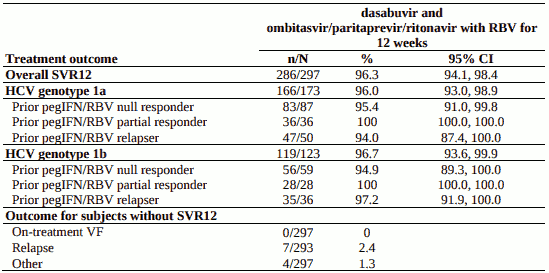
No subjects with HCV genotype 1b infection experienced on-treatment virologic failure and 2 subjects with HCV genotype 1b infection experienced relapse.
PEARL-II - genotype 1b, pegIFN + RBV-experienced, without cirrhosis
Design: randomised, global multicentre, open-label, regimen-controlled
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir without ribavirin or with weight-based ribavirin for 12 weeks
Treated subjects (N=179) had a median age of 57 years (range: 26 to 70); 35.2% were prior pegIFN/RBV null responders; 28.5% were prior pegIFN/RBV partial responders; and 36.3% were prior pegIFN/RBV relapsers; 54.2% were male; 3.9% were Black; ; 12.8% had a history of depression or bipolar disorder; 87.7% had baseline HCV RNA levels of at least 800,000 IU/mL; 17.9% had portal fibrosis (F2) and 14.0% had bridging fibrosis (F3).
Table 12. SVR12 for genotype 1b-infected peginterferon+ribavirin-experienced subjects in PEARL II:
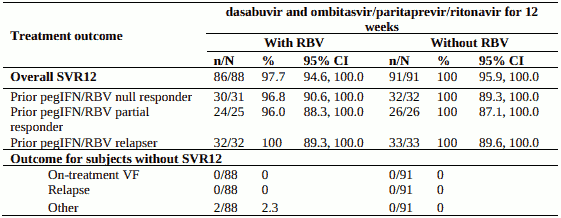
Clinical trial in subjects with compensated cirrhosis
TURQUOISE-II - treatment-naïve or pegIFN + RBV-experienced with compensated cirrhosis
Design: randomised, global multicentre, open-label
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir with weight-based ribavirin for 12 or 24 weeks
Treated subjects (N=380) had a median age of 58 years (range: 21 to 71); 42.1% were treatment-naïve, 36.1% were prior pegIFN/RBV null responders; 8.2% were prior pegIFN/RBV partial responders, 13.7% were prior pegIFN/RBV relapsers; 70.3% were male; 3.2% were Black; 14.7% had platelet counts of less than 90 x 109 /L; 49.7% had albumin less than 40 g/L; 86.1% had baseline HCV RNA levels of at least 800,000 IU/mL; 24.7% had a history of depression or bipolar disorder; 68.7% had HCV genotype 1a infection, 31.3% had HCV genotype 1b infection.
Table 13. SVR12 for genotype 1-infected subjects with compensated cirrhosis who were treatment-naïve or previously treated with pegIFN/RBV:
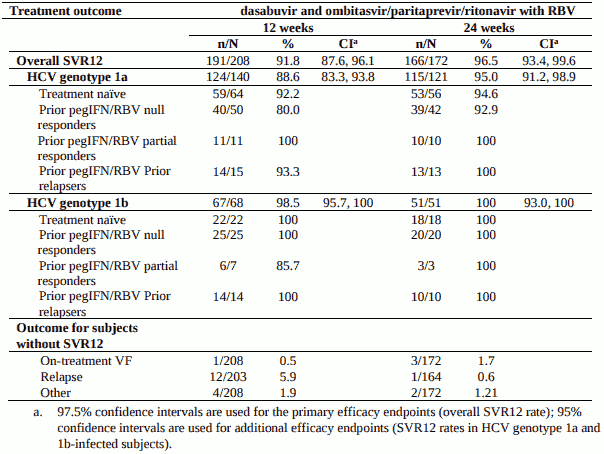
Relapse rates in GT1a cirrhotic subjects by baseline laboratory values are presented in Table 14.
Table 14. TURQUOISE-II: relapse rates by baseline laboratory values after 12 and 24 weeks of treatment in subjects with genotype 1a infection and compensated cirrhosis:
| dasabuvir and ombitasvir/paritaprevir/ritonavir with RBV 12-week arm | dasabuvir and ombitasvir/paritaprevir/ritonavir with RBV 24-week arm | |
|---|---|---|
| Number of Responders at the End of Treatment | 135 | 113 |
| AFP* <20 ng/mL, platelets ≥90 x 109/L, AND albumin ≥35 g/L prior to treatment | ||
| Yes (for all three parameters listed above) | 1/87 (1%) | 0/68 (0%) |
| No (for any parameter listed above) | 10/48 (21%) | 1/45 (2%) |
* AFP= serum alpha fetoprotein
In subjects with all three favourable baseline laboratory values (AFP <20 ng/mL, platelets ≥90 x 109 /L, and albumin ≥35 g/L), relapse rates were similar in subjects treated for 12 or 24 weeks.
TURQUOISE-III: treatment-naïve or pegIFN + RBV-experienced with compensated cirrhosis
Design: global multicentre, open-label
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir without ribavirin for 12 weeks 60 patients were randomized and treated, and 60/60 (100%) achieved SVR12. Main characteristics are shown below.
Table 15. Main demographics in TURQUOISE-III:
| Characteristics | N=60 |
|---|---|
| Age, median (range) years | 60.5 (26-78) |
| Male gender, n (%) | 37 (61) |
| Prior HCV Treatment: | |
| naïve, n (%) | 27 (45) |
| Peg-IFN + RBV, n (%) | 33 (55) |
| Baseline albumin, median g/L | 40.0 |
| <35, n (%) | 10 (17) |
| ≥35, n (%) | 50 (83) |
| Baseline platelet count, median (× 109/L) | 132.0 |
| <90, n (%) | 13 (22) |
| ≥90, n (%) | 47 (78) |
Pooled analyses of clinical trials
Durability of response
Overall, 660 subjects in Phase 2 and 3 clinical trials had HCV RNA results for both the SVR12 and SVR24 time points. Among these subjects, the positive predictive value of SVR12 on SVR24 was 99.8%.
Pooled efficacy analysis
In Phase 3 clinical trials, 1075 subjects (including 181 with compensated cirrhosis) received the recommended regimen (see section 4.2). Table 16 shows SVR rates for these subjects.
In subjects who received the recommended regimen, 97% achieved SVR overall (among which 181 subjects with compensated cirrhosis achieved 97% SVR), while 0.5% experienced virologic breakthrough and 1.2% experienced post-treatment relapse.
Table 16. SVR12 rates for recommended treatment regimens by patient population:

Impact of ribavirin dose adjustment on probability of SVR
In Phase 3 clinical trials, 91.5% of subjects did not require ribavirin dose adjustments during therapy. In the 8.5% of subjects who had ribavirin dose adjustments during therapy, the SVR rate (98.5%) was comparable to subjects who maintained their starting ribavirin dose throughout treatment.
TURQUOISE-I: treatment-naïve or pegIFN + RBV-experienced with HCV GT1 or GT4/HIV-1 coinfection, without cirrhosis or with compensated cirrhosis
Design: randomised, global multicentre, open-label
Treatment: ombitasvir/paritaprevir/ritonavir with or without dasabuvir coadminstered with or without weight-based ribavirin for 12 or 24 weeks
See section 4.2 for dosing recommendations in HCV/HIV-1 co-infected patients. Subjects were on a stable HIV-1 antiretroviral therapy (ART) regimen that included ritonavir-boosted atazanavir or raltegravir, dolutegravir (Part 2 only), or darunavir (Part 1b and Part 2 GT4 only)-, co-administered with a backbone of tenofovir plus emtricitabine or lamivudine. Part 1 of the study was a Phase 2 pilot cohort consisting of 2 parts, Part 1a (63 subjects) and Part 1b (22 subjects). Part 2 was a Phase 3 cohort consisting of 233 subjects.
In Part 1a, all subjects received dasabuvir and ombitasvir/paritaprevir/ritonavir with ribavirin for 12 or 24 weeks. Treated subjects (N = 63) had a median age of 51 years (range: 31 to 69); 24% were Black; 19% had compensated cirrhosis; 67% were treatment-naïve; 33% had failed prior treatment with pegIFN/RBV; 89% had HCV genotype 1a infection.
In Part 1b, all subjects received dasabuvir and ombitasvir/paritaprevir/ritonavir with ribavirin for 12 weeks. Treated subjects (N = 22) had a median age of 54 years (range: 34 to 68); 41% were Black; 14% had compensated cirrhosis; 86% were HCV treatment-naïve; 14% had failed prior treatment with pegIFN/RBV; 68% had HCV genotype 1a infection.
In Part 2, subjects with HCV GT1 received dasabuvir and ombitasvir/paritaprevir/ritonavir with or without ribavirin for 12 or 24 weeks. Subjects with HCV GT4 received ombitasvir/paritaprevir/ritonavir with ribavirin for 12 or 24 Weeks. Treated subjects (N = 233) had a median age of 49 years (range: 26 to 69); 10% were Black; 12% had compensated cirrhosis; 66% were treatment-naïve; 32% had failed prior treatment with pegIFN/RBV; 2% had failed prior treatment with sofosbuvir.
Table 17 shows the primary efficacy analysis of SVR12 performed on subjects with HCV GT1/HIV-1 co-infection that received recommended regimen in Part 2 of the TURQUOISE-I study.
Table 17. Primary SVR12 Assessment for Part 2, Subjects with HCV GT1/HIV-1 co-infection in TURQUOISE-I:

Efficacy analyses performed on other parts of the study demonstrated similarly high SVR12 rates. In Part 1a, SVR12 was achieved by 29/31 (93.5%) subjects on the 12-week arm (95% CI: 79.3% – 98.2%) and by 29/32 (90.6%) subjects on the 24-week arm (95% CI: 75.8% – 96.8%). There was 1 relapse in the 12-week arm and 1 on-treatment virologic failure in the 24-week arm. In Part 1b, SVR12 was achieved by 22/22 (100%) subjects (95% CI: 85.1%, 100%). In Part 2, SVR12 was achieved by 27/28 (96.4%) subjects with HCV GT4/HIV-1 coinfection (95% CI: 82.3%, 99.4%) with no virologic failures.
The SVR12 rates in HCV/HIV-1 co-infected subjects were thus consistent with SVR12 rates in the phase 3 trials of HCV mono-infected subjects.
CORAL-I: treatment-naïve or pegIFN + RBV-experienced, GT 1 or GT4 infected, at least 3 months post liver transplant or 12 months post renal transplant
Design: randomised, global multicentre, open-label
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir for 12 or 24 weeks with or without ribavirin (investigator chosen dose) for GT1 and GT4 infection
In subjects with liver transplant, no cirrhosis and GT1 infection, patients were dosed with dasabuvir and ombitasvir/paritaprevir/ritonavir for 12-24 weeks, with and without RBV. Liver transplant subjects with cirrhosis were dosed with dasabuvir and ombitasvir/paritaprevir/ritonavir with RBV (GT1a for 24 weeks [n=4], GT1b for 12 weeks [n=2]). Subjects with renal transplant and no cirrhosis were dosed for 12 weeks (with RBV for GT1a [n=9], without RBV for GT1b [n=3]). Subjects with liver transplant and GT4 infection were dosed with ombitasvir/paritaprevir/ritonavir with RBV (noncirrhotic for 12 weeks [n=2] and cirrhotic for 24 weeks [n=1]. The dose of ribavirin was individualized at the discretion of the investigator, with most subjects receiving 600 to 800 mg as a starting dose, and most subjects also receiving 600 to 800 mg per day at the end of treatment.
A total of 129 subjects were treated, 84 with GT1a, 41 with GT1b, 1 with GT1 other, 3 with GT4 infection. Overall, 61% had fibrosis stage F0-F1, 26% F2, 9% F3, and 4% F4. 61% had prior HCV treatment experience before transplant. For immunosuppressive medication, most subjects were taking tacrolimus (81%), with the remainder taking cyclosporine.
Among all GT1 subjects who were post liver transplant, 111/114 (97.4%) achieved SVR12; with 2 relapsing post treatment and 1 breakthrough on treatment. Among the GT1 subjects who were post renal transplant, 9/12 (75%) achieved SVR12; however, there were no virologic failures. All 3 (100%) subjects with GT 4 infection who were post liver transplant achieved SVR12
Clinical Trial in patients receiving chronic opioid substitution therapy
In a phase 2, multicentre, open-label, single arm study, 38 treatment-naïve or pegIFN/RBV treatment experienced, non-cirrhotic subjects with genotype 1 infection who were on stable doses of methadone (N=19) or buprenorphine with or without naloxone (N=19) received 12 weeks of dasabuvir in combination with ombitasvir/paritaprevir/ritonavir and ribavirin. Treated subjects had a median age of 51 years (range: 26 to 64); 65.8% were male and 5.3% were Black. A majority (86.8%) had baseline HCV RNA levels of at least 800,000 IU/mL and a majority (84.2%) had genotype 1a infection; 15.8% had portal fibrosis (F2) and 5.3% had bridging fibrosis (F3); and 94.7% were naïve to prior HCV treatment.
Overall, 37 (97.4%) of 38 subjects achieved SVR12. No subjects experienced on-treatment virologic failure or relapse.
RUBY-I; treatment-naïve or pegIFN + RBV experienced with or without cirrhosis who have severe renal impairment or end stage renal disease (ESRD)
Design: multicentre, open-label
Treatment: dasabuvir and ombitasvir/paritaprevir/ritonavir with or without RBV for 12 or 24 weeks
Severe renal impairment or ESRD includes CKD Stage 4 defined as eGFR <30-15 mL/min/1.73 m² or CKD Stage 5 defined as <15 mL/min/1.73 m² or requiring haemodialysis. Treated subjects (N=68) had a median age of 58 years (range: 32-77 years); 83.8% were male; 58.8% were Black; 73.5% of subjects were infected with HCV GT1a; 75.0%% had Stage 5 CKD and 69.1% were on haemodialysis.
Sixty four of 68 (94.1%) subjects achieved SVR12. One subject experienced relapse at Post-Treatment Week 4, 2 subjects prematurely discontinued study drug and 1 subject had missing SVR12 data.
See also Section 4.8 for discussion of safety information for RUBY-I.
In another open-label phase 3b study evaluating 12 weeks of ombitasvir/paritaprevir/ritonavir with or without dasabuvir and without RBV in non-cirrhotic, treatment-naive GT1a and GT4 patients with CKD stage 4 or 5 (Ruby II), the SVR12 rate was 94.4% (17/18), with no subjects experiencing ontreatment virologic failure or relapse.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with dasabuvir and ombitasvir/paritaprevir/ritonavir in one or more subsets of the paediatric populations in the treatment of chronic hepatitis C (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
The pharmacokinetic properties of the combination of dasabuvir with ombitasvir/paritaprevir/ritonavir have been evaluated in healthy adult subjects and in subjects with chronic hepatitis C. Table 18 shows mean Cmax and AUC of dasabuvir 250 mg twice daily with ombitasvir/paritaprevir/ritonavir 25 mg/150 mg/100 mg once daily following multiple doses with food in healthy volunteers.
Table 18. Geometric mean Cmax, AUC of multiple doses of dasabuvir 250 mg twice daily and ombitasvir/paritaprevir/ritonavir 25 mg/150 mg/100 mg once daily with food in healthy volunteers:
| Cmax (ng/ml) (CV%) | AUC (ng*hr/ml) (CV%) | |
|---|---|---|
| Dasabuvir | 1030 (31) | 6840 (32) |
Absorption
Dasabuvir was absorbed after oral administration with mean Tmax of approximately 4 to 5 hours. Dasabuvir exposures increased in a dose proportional manner and accumulation is minimal. Pharmacokinetic steady state for dasabuvir when coadministered with ombitasvir/paritaprevir/ritonavir is achieved after approximately 12 days of dosing.
Effects of food
Dasabuvir should be administered with food. All clinical trials with dasabuvir have been conducted following administration with food.
Food increased the exposure (AUC) of dasabuvir by up to 30% relative to the fasting state. The increase in exposure was similar regardless of meal type (e.g., high-fat versus moderate-fat) or calorie content (approximately 600 kcal versus approximately 1000 kcal). To maximise absorption, dasabuvir should be taken with food without regard to fat or calorie content.
Distribution
Dasabuvir is highly bound to plasma proteins. Plasma protein binding is not meaningfully altered in patients with renal or hepatic impairment. The blood to plasma concentration ratios in human ranged from 0.5 to 0.7 indicating that dasabuvir was preferentially distributed in the plasma compartment of whole blood. Dasabuvir was greater than 99.5%, and M1 major metabolite of dasabuvir was 94.5% bound to human plasma proteins over a concentration range of 0.05 to 5 µg/mL. At steady-state the exposures ratio of M1 to dasabuvir is approximately 0.6. Taking into account the protein binding and in vitro activity of M1 against HCV genotype 1, its contribution to efficacy is expected to be similar to that of dasabuvir. In addition, M1 is a substrate of the hepatic uptake transporters OATP family and OCT1 and thus, the hepatocyte concentration and thereby contribution to efficacy, may be larger than dasabuvir.
Biotransformation
Dasabuvir is predominantly metabolised by CYP2C8 and to a lesser extent by CYP3A. Following a 400 mg 14C-dasabuvir dose in humans, unchanged dasabuvir was the major component (approximately 60%) of drug related radioactivity in plasma. Seven metabolites were identified in plasma. The most abundant plasma metabolite was M1, which represented 21% of drug-related radioactivity (AUC) in circulation following single dose; it's formed via oxidative metabolism predominantly by CYP2C8.
Elimination
Following dosing of dasabuvir with ombitasvir/paritaprevir/ritonavir, mean plasma half-life of dasabuvir was approximately 6 hours. Following a 400 mg 14C-dasabuvir dose, approximately 94% of the radioactivity was recovered in faeces with limited radioactivity (approximately 2%) in urine. Unchanged dasabuvir accounted for 26.2% and M1 for 31.5% of the total dose in faeces. M1 is mainly cleared through direct biliary excretion with the contribution of UGT-mediated glucuronidation and, to a small extent, oxidative metabolism.
Dasabuvir does not inhibit organic anion transporter (OAT1) in vivo and is not expected to inhibit organic cation transporters (OCT2), organic anion transporters (OAT3), or multidrug and toxin extrusion proteins (MATE1 and MATE2K) at clinically relevant concentrations; therefore, dasabuvir does not affect medicinal product transport by these proteins.
Special populations
Elderly
Based on population pharmacokinetic analysis of data from Phase 3 clinical studies, a 10 year increase or decrease in age from 54 years (median age in the Phase 3 studies) would results in <10% change in dasabuvir exposures. There is no pharmacokinetic information in patients >75 years.
Sex or body weight
Based on population pharmacokinetic analysis of data from Phase 3 clinical studies, female subjects would have approximately 14 to 30% higher dasabuvir exposures than male subjects. A 10 kg change in body weight from 76 kg (median weight in the Phase 3 studies) would result in <10% change in dasabuvir exposures.
Race or ethnicity
Based on population pharmacokinetic analysis of data from Phase 3 clinical studies, Asian subjects had 29% to 39% higher dasabuvir exposures than non-Asian subjects.
Renal impairment
Pharmacokinetics of the combination of ombitasvir 25 mg, paritaprevir 150 mg, and ritonavir 100 mg, with dasabuvir 400 mg were evaluated in subjects with mild (CrCl: 60 to 89 ml/min), moderate (CrCl: 30 to 59 ml/min) and severe (CrCl: 15 to 29 ml/min) renal impairment, relative to subjects with normal renal function.
In subjects with mild, moderate and severe renal impairment, dasabuvir mean AUC values were 21% higher, 37% higher and 50% higher, respectively. Dasabuvir M1 AUC values were 6% lower, 10% lower, and 13% lower, respectively.
The changes in dasabuvir exposures in subjects with mild, moderate and severe renal impairment are not considered to be clinically significant. Limited data in patients with end-stage renal disease indicate no clinically significant changes in exposure also in this patient group. No dose adjustment of dasabuvir is required for patients with mild, moderate, or severe renal impairment, or end-stage-renal disease on dialysis (see section 4.2).
Hepatic impairment
Pharmacokinetics of the combination of dasabuvir 400 mg, with ombitasvir 25 mg, paritaprevir 200 mg, and ritonavir 100 mg were evaluated in subjects with mild (Child-Pugh A), moderate (ChildPugh B) and severe (Child-Pugh C) hepatic impairment, relative to subjects with normal hepatic function.
In subjects with mild, moderate and severe hepatic impairment, dasabuvir AUC values were 17% higher, 16% lower and 325% higher, respectively. The AUC values of dasabuvir M1 metabolite were unchanged, 57% lower, and 77% higher, respectively. Plasma protein binding of dasabuvir and its M1 metabolite were not meaningfully different in subjects with hepatic impairment compared to normal control subjects (see sections 4.2, 4.4 and 4.8).
Paediatric population
The pharmacokinetics of dasabuvir with ombitasvir/paritaprevir/ritonavir in paediatric patients has not been investigated (see section 4.2).
Preclinical safety data
Dasabuvir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes and in vivo rat micronucleus assays.
Dasabuvir was not carcinogenic in a 6-month transgenic mouse study up to the highest dosage tested (2 g/kg/day), resulting in dasabuvir AUC exposures approximately 19-fold higher than those in humans at the recommended dose of 500 mg (250 mg twice daily).
Similarly, dasabuvir was not carcinogenic in a 2-year rat study up to the highest dose tested (800 mg/kg/day), resulting in dasabuvir exposures approximately 19-fold higher than those in humans at 500 mg.
Dasabuvir had no effects on embryo-foetal viability or on fertility in rodents and were not teratogenic in two species. No adverse effects on behaviour, reproduction or development of offspring were reported. The highest dasabuvir dose tested produced exposures equal to 16 to 24-fold (rat) or 6-fold (rabbit) the exposures in humans at the maximum recommended clinical dose.
Dasabuvir was the predominant component observed in the milk of lactating rats, without effect on nursing pups. Elimination half-life in rat milk was slightly shorter than in plasma, AUC was about 2 fold of that in plasma. Since dasabuvir is a BCRP substrate, distribution to the milk may change if this transporter is inhibited or induced by co-administration of other medicinal products. Dasabuvirderived material was minimally transferred through the placenta in pregnant rats.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.