FAMPYRA Prolonged-release tablet Ref.[8707] Active ingredients: Fampridine
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Biogen Netherlands B.V., Prins Mauritslaan 13, 1171 LP, Badhoevedorp, The Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: Other nervous system drugs
ATC code: N07XX07
Pharmacodynamic effects
Fampyra is a potassium channel blocker. By blocking potassium channels, Fampyra reduces the leakage of ionic current through these channels, thereby prolonging repolarization and thus enhancing action potential formation in demyelinated axons and neurological function. Presumably, by enhancing action potential formation, more impulses might be conducted in the central nervous system.
Clinical efficacy and safety
Three phase III, randomised, double-blind, placebo controlled confirmatory studies, (MS-F203 and MS-F204 and 218MS305) have been performed. The proportion of responders was independent of concomitant immunomodulatory therapy (including interferons, glatiramer acetate, fingolimod and natalizumab). The Fampyra dose was 10 mg BID.
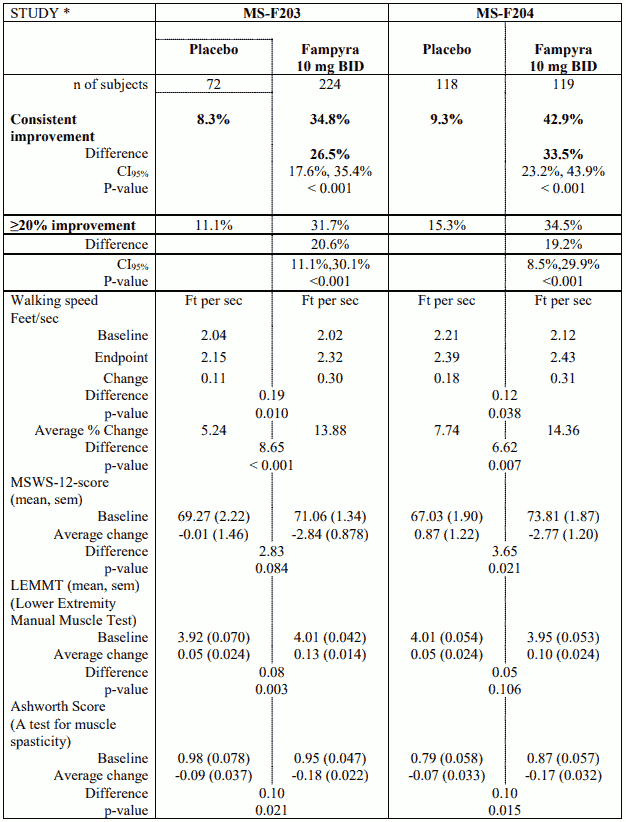
Studies MS-F203 and MS-F204
The primary endpoint in studies MS-F203 and MS-F204 was the responder rate in walking speed as measured by the Timed 25-foot Walk (T25FW). A responder was defined as a patient who consistently had a faster walking speed for at least three visits out of a possible four during the double blind period as compared to the maximum value among five off-treatment visits.
A significantly greater proportion of Fampyra treated patients were responders as compared to placebo (MS-F203: 34.8% vs. 8.3%, p<0.001; MS-F204: 42.9% vs. 9.3%, p<0.001).
Patients who responded to Fampyra increased their walking speed on average by 26.3% vs 5.3% on placebo (p<0.001) (MS-F203) and 25.3% vs 7.8% (p< 0.001) (MS-F204). The improvement appeared rapidly (within weeks) after starting Fampyra.
Statistically and clinically meaningful improvements in walking were seen, as measured by the 12-item Multiple Sclerosis Walking Scale.
Table 1. Studies MS-F203 and MS-F204:
Study 218MS305
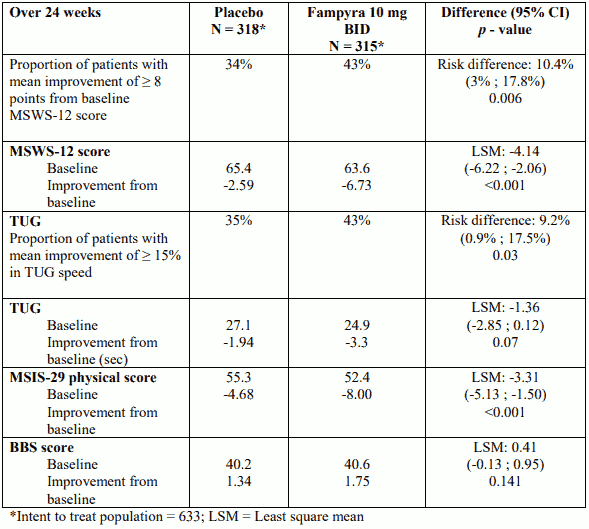
Study 218MS305 was conducted in 636 subjects with multiple sclerosis and walking disability. Duration of double-blind treatment was 24 weeks with a 2 week post–treatment follow-up. The primary endpoint was improvement in walking ability, measured as the proportion of patients achieving a mean improvement of ≥ 8 points from baseline MSWS-12 score over 24 weeks. In this study there was a statistically significant treatment difference, with a greater proportion of Fampyra treated patients demonstrating an improvement in walking ability, compared to placebo-controlled patients (relative risk of 1.38 (95% CI: [1.06, 1.70]). Improvements generally appeared within 2 to 4 weeks of initiation of treatment, and disappeared within 2 weeks of treatment cessation.
Fampyra treated patients also demonstrated a statistically significant improvement in the Timed Up and Go (TUG) test, a measure of static and dynamic balance and physical mobility. In this secondary endpoint, a greater proportion of Fampyra treated patients achieved ≥15% mean improvement from baseline TUG speed over a 24 week period, compared to placebo. The difference in the Berg Balance Scale (BBS; a measure of static balance), was not statistically significant.
In addition, patients treated with Fampyra demonstrated a statistically significant mean improvement from baseline compared to placebo in the Multiple Sclerosis Impact Scale (MSIS-29) physical score (LSM difference -3.31, p<0.001).
Table 2. Study 218MS305:
The European Medicines Agency has waived the obligation to submit the results of studies with Fampyra in all subsets of the paediatric population in treatment of multiple sclerosis with walking disability (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Orally administered fampridine is rapidly and completely absorbed from the gastrointestinal tract. Fampridine has a narrow therapeutic index. Absolute bioavailability of Fampyra prolonged-release tablets has not been assessed, but relative bioavailability (as compared to an aqueous oral solution) is 95%. The Fampyra prolonged-release tablet has a delay in the absorption of fampridine manifested by slower rise to a lower peak concentration, without any effect on the extent of absorption.
When Fampyra tablets are taken with food, the reduction in the area under the plasma concentrationtime curve (AUC0-∞) of fampridine is approximately 2-7% (10 mg dose). The small reduction in AUC is not expected to cause a reduction in the therapeutic efficacy. However, Cmax increases by 15-23%. Since there is a clear relationship between Cmax and dose related adverse reactions, it is recommended to take Fampyra without food (see section 4.2).
Distribution
Fampridine is a lipid-soluble medicinal product which readily crosses the blood-brain barrier. Fampridine is largely unbound to plasma proteins (bound fraction varied between 3-7% in human plasma). Fampridine has a volume of distribution of approximately 2.6 l/kg. Fampridine is not a substrate for P-glycoprotein.
Biotransformation
Fampridine is metabolised in humans by oxidation to 3-hydroxy-4-aminopyridine and further conjugated to the 3-hydroxy-4-aminopyridine sulfate. No pharmacological activity was found for the fampridine metabolites against selected potassium channels in vitro.
The 3-hydroxylation of fampridine to 3-hydroxy-4-aminopyridine by human liver microsomes appeared to be catalyzed by Cytochrome P450 2E1 (CYP2E1).
There was evidence of direct inhibition of CYP2E1 by fampridine at 30 μM (approximately 12% inhibition) which is approximately 100 times the average plasma fampridine concentration measured for the 10 mg tablet.
Treatment of cultured human hepatocytes with fampridine had little or no effect on induction of CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2E1 or CYP3A4/5 enzyme activities.
Elimination
The major route of elimination for fampridine is renal excretion, with approximately 90% of the dose recovered in urine as parent medicinal product within 24 hours. Renal clearance (CLR 370 ml/min) is substantially greater than glomerular filtration rate due to combined glomerular filtration and active excretion by the renal OCT2 transporter. Faecal excretion accounts for less than 1% of the administered dose.
Fampyra is characterized by linear (dose-proportional) pharmacokinetics with a terminal elimination half-life of approximately 6 hours. The maximum plasma concentration (Cmax) and, to a smaller extent, area under the plasma concentration-time curve (AUC) increase proportionately with dose. There is no evidence of clinically relevant accumulation of fampridine taken at the recommended dose in patients with full renal function. In patients with renal impairment, accumulation occurs relative to the degree of impairment.
Special Populations
Older people
Clinical studies of Fampyra did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger patients. Fampyra is primarily excreted unchanged by the kidneys, and with creatinine clearance known to decrease with age, monitoring of renal function in older patients should be considered (see section 4.2).
Paediatric Population
No data are available.
Patients with renal impairment
Fampridine is eliminated primarily by the kidneys as unchanged medicinal product and therefore renal function should be checked in patients where renal function might be compromised. Patients with mild renal impairment can be expected to have approximately 1.7 to 1.9 times the fampridine concentrations achieved by patients with normal renal function. Fampyra must not be administered to patients with mild, moderate and severe renal impairment (see section 4.3).
Preclinical safety data
Fampridine was studied in oral repeat dose toxicity studies in several animal species.
Adverse responses to orally administered fampridine were rapid in onset, most often occurring within the first 2 hours post-dose. Clinical signs evident after large single doses or repeated lower doses were similar in all species studied and included tremors, convulsions, ataxia, dyspnoea, dilated pupils, prostration, abnormal vocalization, increased respiration, and excess salivation. Gait abnormalities and hyper-excitability were also observed. These clinical signs were not unexpected and represent exaggerated pharmacology of fampridine. In addition, single cases of fatal urinary tract obstructions were observed in rats. The clinical relevance of these findings remains to be elucidated, but a causal relationship with fampridine treatment cannot be excluded.
In reproduction toxicity studies in rats and rabbits, decreased weight and viability of foetuses and offspring were observed at maternally toxic doses. However, no increased risk for malformations or adverse effects on fertility was noted.
In a battery of in vitro and in vivo studies fampridine did not show any potential to be mutagenic, clastogenic or carcinogenic.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.