GILVEC Film-coated tablet Ref.[8027] Active ingredients: Imatinib
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
Pharmacodynamic properties
Pharmacotherapeutic group: protein-tyrosine kinase inhibitor
ATC code: L01XE01
Mechanism of action
Imatinib is a small molecule protein-tyrosine kinase inhibitor that potently inhibits the activity of the Bcr-Abl tyrosine kinase (TK), as well as several receptor TKs: Kit, the receptor for stem cell factor (SCF) coded for by the c-Kit proto-oncogene, the discoidin domain receptors (DDR1 and DDR2), the colony stimulating factor receptor (CSF-1R) and the platelet-derived growth factor receptors alpha and beta (PDGFR-alpha and PDGFR-beta). Imatinib can also inhibit cellular events mediated by activation of these receptor kinases.
Pharmacodynamic effects
Imatinib is a protein-tyrosine kinase inhibitor which potently inhibits the Bcr-Abl tyrosine kinase at the in vitro, cellular and in vivo levels. The compound selectively inhibits proliferation and induces apoptosis in Bcr-Abl positive cell lines as well as fresh leukaemic cells from Philadelphia chromosome positive CML and acute lymphoblastic leukaemia (ALL) patients.
In vivo the compound shows anti-tumour activity as a single agent in animal models using Bcr-Abl positive tumour cells.
Imatinib is also an inhibitor of the receptor tyrosine kinases for platelet-derived growth factor (PDGF), PDGF-R, and stem cell factor (SCF), c-Kit, and inhibits PDGF- and SCF-mediated cellular events. In vitro, imatinib inhibits proliferation and induces apoptosis in gastrointestinal stromal tumour (GIST) cells, which express an activating kit mutation. Constitutive activation of the PDGF receptor or the Abl protein tyrosine kinases as a consequence of fusion to diverse partner proteins or constitutive production of PDGF have been implicated in the pathogenesis of MDS/MPD, HES/CEL and DFSP. Imatinib inhibits signalling and proliferation of cells driven by dysregulated PDGFR and Abl kinase activity.
Clinical studies in chronic myeloid leukaemia
The effectiveness of Glivec is based on overall haematological and cytogenetic response rates and progression-free survival. Except in newly diagnosed chronic phase CML, there are no controlled trials demonstrating a clinical benefit, such as improvement in disease-related symptoms or increased survival.
Three large, international, open-label, non-controlled phase II studies were conducted in patients with Philadelphia chromosome positive (Ph+) CML in advanced, blast or accelerated phase disease, other Ph+ leukaemias or with CML in the chronic phase but failing prior interferon-alpha (IFN) therapy. One large, open-label, multicentre, international randomised phase III study has been conducted in patients with newly diagnosed Ph+ CML. In addition, children have been treated in two phase I studies and one phase II study.
In all clinical studies 38-40% of patients were ≥60 years of age and 10-12% of patients were ≥70 years of age.
Chronic phase, newly diagnosed: This phase III study in adult patients compared treatment with either single-agent Glivec or a combination of interferon-alpha (IFN) plus cytarabine (Ara-C). Patients showing lack of response (lack of complete haematological response (CHR) at 6 months, increasing WBC, no major cytogenetic response (MCyR) at 24 months), loss of response (loss of CHR or MCyR) or severe intolerance to treatment were allowed to cross over to the alternative treatment arm. In the Glivec arm, patients were treated with 400 mg daily. In the IFN arm, patients were treated with a target dose of IFN of 5 MIU/m²/day subcutaneously in combination with subcutaneous Ara-C 20 mg/m²/day for 10 days/month.
A total of 1,106 patients were randomised, 553 to each arm. Baseline characteristics were well balanced between the two arms. Median age was 51 years (range 18-70 years), with 21.9% of patients ≥60 years of age. There were 59% males and 41% females; 89.9% caucasian and 4.7% black patients. Seven years after the last patient had been recruited, the median duration of first-line treatment was 82 and 8 months in the Glivec and IFN arms, respectively. The median duration of second-line treatment with Glivec was 64 months. Overall, in patients receiving first-line Glivec, the average daily dose delivered was 406 ± 76 mg. The primary efficacy endpoint of the study is progression-free survival. Progression was defined as any of the following events: progression to accelerated phase or blast crisis, death, loss of CHR or MCyR, or in patients not achieving a CHR an increasing WBC despite appropriate therapeutic management. Major cytogenetic response, haematological response, molecular response (evaluation of minimal residual disease), time to accelerated phase or blast crisis and survival are main secondary endpoints. Response data are shown in Table 2.
Table 2. Response in newly diagnosed CML Study (84-month data):
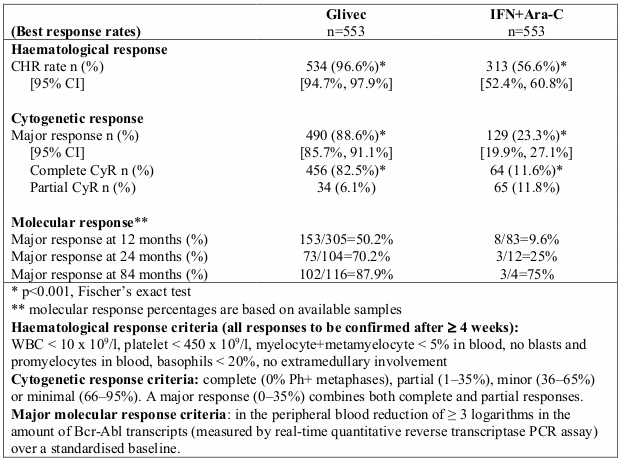
Rates of complete haematological response, major cytogenetic response and complete cytogenetic response on first-line treatment were estimated using the Kaplan-Meier approach, for which non- responses were censored at the date of last examination. Using this approach, the estimated cumulative response rates for first-line treatment with Glivec improved from 12 months of therapy to 84 months of therapy as follows: CHR from 96.4% to 98.4% and CCyR from 69.5% to 87.2%, respectively.
With 7 years follow-up, there were 93 (16.8%) progression events in the Glivec arm: 37 (6.7%) involving progression to accelerated phase/blast crisis, 31 (5.6%) loss of MCyR, 15 (2.7%) loss of CHR or increase in WBC, and 10 (1.8%) CML unrelated deaths. In contrast, there were 165 (29.8%) events in the IFN+Ara-C arm, of which 130 occurred during first-line treatment with IFN+Ara-C.
The estimated rate of patients free of progression to accelerated phase or blast crisis at 84 months was significantly higher in the Glivec arm compared to the IFN arm (92.5% versus 85.1%, p<0.001). The annual rate of progression to accelerated phase or blast crisis decreased with time on therapy and was less than 1% annually in the fourth and fifth years. The estimated rate of progression-free survival at 84 months was 81.2% in the Glivec arm and 60.6% in the control arm (p<0.001). The yearly rates of progression of any type for Glivec also decreased over time.
A total of 71 (12.8%) and 85 (15.4%) patients died in the Glivec and IFN+Ara-C groups, respectively. At 84 months the estimated overall survival is 86.4% (83, 90) vs. 83.3% (80, 87) in the randomised Glivec and the IFN+Ara-C groups, respectively (p=0.073, log-rank test). This time-to-event endpoint is strongly affected by the high crossover rate from IFN+Ara-C to Glivec. The effect of Glivec treatment on survival in chronic phase, newly diagnosed CML has been further examined in a retrospective analysis of the above reported Glivec data with the primary data from another Phase III study using IFN+Ara-C (n=325) in an identical regimen. In this retrospective analysis, the superiority of Glivec over IFN+Ara-C in overall survival was demonstrated (p<0.001); within 42 months, 47 (8.5%) Glivec patients and 63 (19.4%) IFN+Ara-C patients had died.
The degree of cytogenetic response and molecular response had a clear effect on long-term outcomes in patients on Glivec. Whereas an estimated 96% (93%) of patients with CCyR (PCyR) at 12 months were free of progression to accelerated phase/blast crisis at 84 months, only 81% of patients without MCyR at 12 months were free of progression to advanced CML at 84 months (p<0.001 overall, p=0.25 between CCyR and PCyR). For patients with reduction in Bcr-Abl transcripts of at least 3 logarithms at 12 months, the probability of remaining free from progression to accelerated phase/blast crisis was 99% at 84 months. Similar findings were found based on a 18-months landmark analysis.
In this study, dose escalations were allowed from 400 mg daily to 600 mg daily, then from 600 mg daily to 800 mg daily. After 42 months of follow-up, 11 patients experienced a confirmed loss (within 4 weeks) of their cytogenetic response. Of these 11 patients, 4 patients escalated up to 800 mg daily, 2 of whom regained a cytogenetic response (1 partial and 1 complete, the latter also achieving a molecular response), while of the 7 patients who did not escalate the dose, only one regained a complete cytogenetic response. The percentage of some adverse reactions was higher in the 40 patients in whom the dose was increased to 800 mg daily compared to the population of patients before dose increase (n=551). The more frequent adverse reactions included gastrointestinal haemorrhages, conjunctivitis and elevation of transaminases or bilirubin. Other adverse reactions were reported with lower or equal frequency.
Chronic phase, Interferon failure
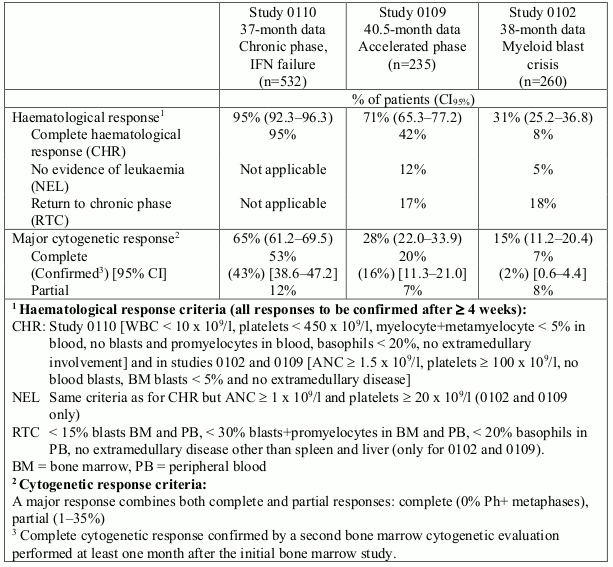
532 adult patients were treated at a starting dose of 400 mg. The patients were distributed in three main categories: haematological failure (29%), cytogenetic failure (35%), or intolerance to interferon (36%). Patients had received a median of 14 months of prior IFN therapy at doses ≥25 x 106 IU/week and were all in late chronic phase, with a median time from diagnosis of 32 months. The primary efficacy variable of the study was the rate of major cytogenetic response (complete plus partial response, 0 to 35% Ph+ metaphases in the bone marrow).
In this study 65% of the patients achieved a major cytogenetic response that was complete in 53% (confirmed 43%) of patients (Table 3). A complete haematological response was achieved in 95% of patients.
Accelerated phase
235 adult patients with accelerated phase disease were enrolled. The first 77 patients were started at 400 mg, the protocol was subsequently amended to allow higher dosing and the remaining 158 patients were started at 600 mg.
The primary efficacy variable was the rate of haematological response, reported as either complete haematological response, no evidence of leukaemia (i.e. clearance of blasts from the marrow and the blood, but without a full peripheral blood recovery as for complete responses), or return to chronic phase CML. A confirmed haematological response was achieved in 71.5% of patients (Table 3). Importantly, 27.7% of patients also achieved a major cytogenetic response, which was complete in 20.4% (confirmed 16%) of patients. For the patients treated at 600 mg, the current estimates for median progression-free-survival and overall survival were 22.9 and 42.5 months, respectively.
Myeloid blast crisis
260 patients with myeloid blast crisis were enrolled. 95 (37%) had received prior chemotherapy for treatment of either accelerated phase or blast crisis ("pretreated patients") whereas 165 (63%) had not ("untreated patients"). The first 37 patients were started at 400 mg, the protocol was subsequently amended to allow higher dosing and the remaining 223 patients were started at 600 mg.
The primary efficacy variable was the rate of haematological response, reported as either complete haematological response, no evidence of leukaemia, or return to chronic phase CML using the same criteria as for the study in accelerated phase. In this study, 31% of patients achieved a haematological response (36% in previously untreated patients and 22% in previously treated patients). The rate of response was also higher in the patients treated at 600 mg (33%) as compared to the patients treated at 400 mg (16%, p=0.0220). The current estimate of the median survival of the previously untreated and treated patients was 7.7 and 4.7 months, respectively.
Lymphoid blast crisis
A limited number of patients were enrolled in phase I studies (n=10). The rate of haematological response was 70% with a duration of 2-3 months.
Paediatric patients
A total of 26 paediatric patients of age <18 years with either chronic phase CML (n=11) or CML in blast crisis or Ph+ acute leukaemias (n=15) were enrolled in a dose-escalation phase I trial. This was a population of heavily pretreated patients, as 46% had received prior BMT and 73% a prior multi-agent chemotherapy. Patients were treated at doses of Glivec of 260 mg/m²/day (n=5), 340 mg/m²/day (n=9), 440 mg/m²/day (n=7) and 570 mg/m²/day (n=5). Out of 9 patients with chronic phase CML and cytogenetic data available, 4 (44%) and 3 (33%) achieved a complete and partial cytogenetic response, respectively, for a rate of MCyR of 77%.
A total of 51 paediatric patients with newly diagnosed and untreated CML in chronic phase have been enrolled in an open-label, multicentre, single-arm phase II trial. Patients were treated with Glivec 340 mg/m²/day, with no interruptions in the absence of dose limiting toxicity. Glivec treatment induces a rapid response in newly diagnosed paediatric CML patients with a CHR of 78% after 8 weeks of therapy. The high rate of CHR is accompanied by the development of a complete cytogenetic response (CCyR) of 65% which is comparable to the results observed in adults. Additionally, partial cytogenetic response (PCyR) was observed in 16% for a MCyR of 81%. The majority of patients who achieved a CCyR developed the CCyR between months 3 and 10 with a median time to response based on the Kaplan-Meier estimate of 5.6 months.
The European Medicines Agency has waived the obligation to submit the results of studies with Glivec in all subsets of the paediatric population in Philadelphia chromosome (bcr-abl translocation)- positive chronic myeloid leukaemia (see section 4.2 for information on paediatric use).
Clinical studies in Ph+ ALL
Newly diagnosedPh+ ALL
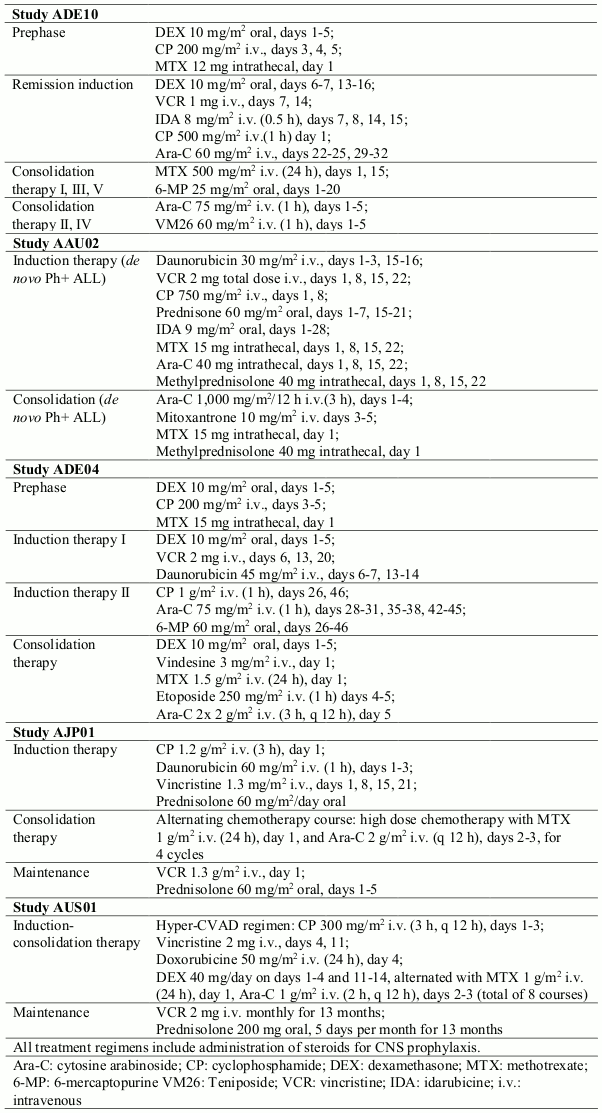
In a controlled study (ADE10) of imatinib versus chemotherapy induction in 55 newly diagnosed patients aged 55 years and over, imatinib used as single agent induced a significantly higher rate of complete haematological response than chemotherapy (96.3% vs. 50%; p=0.0001). When salvage therapy with imatinib was administered in patients who did not respond or who responded poorly to chemotherapy, it resulted in 9 patients (81.8%) out of 11 achieving a complete haematological response. This clinical effect was associated with a higher reduction in bcr- abl transcripts in the imatinib-treated patients than in the chemotherapy arm after 2 weeks of therapy (p=0.02). All patients received imatinib and consolidation chemotherapy (see Table 4) after induction and the levels of bcr-abl transcripts were identical in the two arms at 8 weeks. As expected on the basis of the study design, no difference was observed in remission duration, disease-free survival or overall survival, although patients with complete molecular response and remaining in minimal residual disease had a better outcome in terms of both remission duration (p=0.01) and disease-free survival (p=0.02).
The results observed in a population of 211 newly diagnosed Ph+ ALL patients in four uncontrolled clinical studies (AAU02, ADE04, AJP01 and AUS01) are consistent with the results described above. Imatinib in combination with chemotherapy induction (see Table 4) resulted in a complete haematological response rate of 93% (147 out of 158 evaluable patients) and in a major cytogenetic response rate of 90% (19 out of 21 evaluable patients). The complete molecular response rate was 48% (49 out of 102 evaluable patients). Disease-free survival (DFS) and overall survival (OS) constantly exceeded 1 year and were superior to historical control (DFS p<0.001; OS p<0.0001) in two studies (AJP01 and AUS01).
Table 4. Chemotherapy regimen used in combination with imatinib:
Paediatric patients
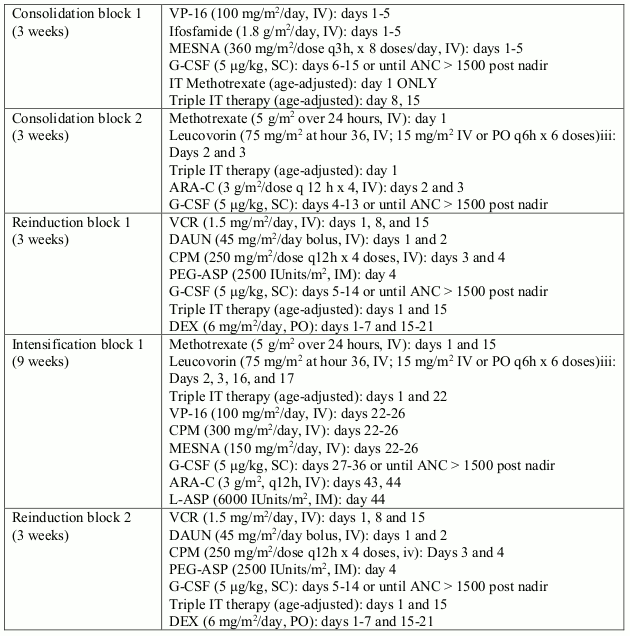
In study I2301, a total of 93 paediatric, adolescent and young adult patients (from 1 to 22 years old) with Ph+ ALL were enrolled in an open-label, multicentre, sequential cohort, non- randomised phase III trial, and were treated with Glivec (340 mg/m²/day) in combination with intensive chemotherapy after induction therapy. Glivec was administered intermittently in cohorts 1-5, with increasing duration and earlier start of Glivec from cohort to cohort; cohort 1 receiving the lowest intensitiy and cohort 5 receiving the highest intensity of Glivec (longest duration in days with continuous daily Glivec dosing during the first chemotherapy treatment courses). Continuous daily exposure to Glivec early in the course of treatment in combination with chemotherapy in cohort 5-patients (n=50) improved the 4-year event-free survival (EFS) compared to historical controls (n=120), who received standard chemotherapy without Glivec (69.6% vs. 31.6%, respectively). The estimated 4-year OS in cohort 5-patients was 83.6% compared to 44.8% in the historical controls. 20 out of the 50 (40%) patients in cohort 5 received haematopoietic stem cell transplant.
Table 5. Chemotherapy regimen used in combination with imatinib in study I2301:
Study AIT07 was a multicentre, open-label, randomised, phase II/III study that included 128 patients (1 to <18 years) treated with imatinib in combination with chemotherapy. Safety data from this study seem to be in line with the safety profile of imatinib in Ph+ ALL patients.
Relapsed/refractory Ph+ ALL
When imatinib was used as single agent in patients with relapsed/refractory Ph+ ALL, it resulted, in the 53 out of 411 patients evaluable for response, in a haematological response rate of 30% (9% complete) and a major cytogenetic response rate of 23%. (Of note, out of the 411 patients, 353 were treated in an expanded access program without primary response data collected.) The median time to progression in the overall population of 411 patients with relapsed/refractory Ph+ ALL ranged from 2.6 to 3.1 months, and median overall survival in the 401 evaluable patients ranged from 4.9 to 9 months. The data was similar when re-analysed to include only those patients age 55 or older.
Clinical studies in MDS/MPD
Experience with Glivec in this indication is very limited and is based on haematological and cytogenetic response rates. There are no controlled trials demonstrating a clinical benefit or increased survival. One open label, multicentre, phase II clinical trial (study B2225) was conducted testing Glivec in diverse populations of patients suffering from life-threatening diseases associated with Abl, Kit or PDGFR protein tyrosine kinases. This study included 7 patients with MDS/MPD who were treated with Glivec 400 mg daily. Three patients presented a complete haematological response (CHR) and one patient experienced a partial haematological response (PHR). At the time of the original analysis, three of the four patients with detected PDGFR gene rearrangements developed haematological response (2 CHR and 1 PHR). The age of these patients ranged from 20 to 72 years.
An observational registry (study L2401) was conducted to collect long-term safety and efficacy data in patients suffering from myeloproliferative neoplasms with PDGFR-β rearrangement and who were treated with Glivec. The 23 patients enrolled in this registry received Glivec at a median daily dose of 264 mg (range: 100 to 400 mg) for a median duration of 7.2 years (range 0.1 to 12.7 years). Due to the observational nature of this registry, haematologic, cytogenetic and molecular assessment data were available for 22, 9 and 17 of the 23 enrolled patients, respectively. When assuming conservatively that patients with missing data were non-responders, CHR was observed in 20/23 (87%) patients, CCyR in 9/23 (39.1%) patients, and MR in 11/23 (47.8%) patients, respectively. When the response rate is calculated from patients with at least one valid assessment, the response rate for CHR, CCyR and MR was 20/22 (90.9%), 9/9 (100%) and 11/17 (64.7%), respectively.
In addition a further 24 patients with MDS/MPD were reported in 13 publications. 21 patients were treated with Glivec 400 mg daily, while the other 3 patients received lower doses. In eleven patients PDGFR gene rearrangements was detected, 9 of them achieved a CHR and 1 PHR. The age of these patients ranged from 2 to 79 years. In a recent publication updated information from 6 of these 11 patients revealed that all these patients remained in cytogenetic remission (range 32-38 months). The same publication reported long term follow-up data from 12 MDS/MPD patients with PDGFR gene rearrangements (5 patients from study B2225). These patients received Glivec for a median of 47 months (range 24 days-60 months). In 6 of these patients follow-up now exceeds 4 years. Eleven patients achieved rapid CHR; ten had complete resolution of cytogenetic abnormalities and a decrease or disappearance of fusion transcripts as measured by RT-PCR. Haematological and cytogenetic responses have been sustained for a median of 49 months (range 19-60) and 47 months (range 16-59), respectively. The overall survival is 65 months since diagnosis (range 25-234). Glivec administration to patients without the genetic translocation generally results in no improvement.
There are no controlled trials in paediatric patients with MDS/MPD. Five (5) patients with MDS/MPD associated with PDGFR gene re-arrangements were reported in 4 publications. The age of these patients ranged from 3 months to 4 years and imatinib was given at dose 50 mg daily or doses ranging from 92.5 to 340 mg/m² daily. All patients achieved complete haematological response, cytogenetic response and/or clinical response.
Clinical studies in HES/CEL
One open-label, multicentre, phase II clinical trial (study B2225) was conducted testing Glivec in diverse populations of patients suffering from life-threatening diseases associated with Abl, Kit or PDGFR protein tyrosine kinases. In this study, 14 patients with HES/CEL were treated with 100 mg to 1,000 mg of Glivec daily. A further 162 patients with HES/CEL, reported in 35 published case reports and case series received Glivec at doses from 75 mg to 800 mg daily. Cytogenetic abnormalities were evaluated in 117 of the total population of 176 patients. In 61 of these 117 patients FIP1L1-PDGFRa fusion kinase was identified. An additional four HES patients were found to be FIP1L1-PDGFRa-positive in other 3 published reports. All 65 FIP1L1-PDGFRa fusion kinase positive patients achieved a CHR sustained for months (range from 1+ to 44+ months censored at the time of the reporting). As reported in a recent publication 21 of these 65 patients also achieved complete molecular remission with a median follow-up of 28 months (range 13-67 months). The age of these patients ranged from 25 to 72 years. Additionally, improvements in symptomatology and other organ dysfunction abnormalities were reported by the investigators in the case reports.
Improvements were reported in cardiac, nervous, skin/subcutaneous tissue, respiratory/thoracic/mediastinal, musculoskeletal/connective tissue/vascular, and gastrointestinal organ systems.
There are no controlled trials in paediatric patients with HES/CEL. Three (3) patients with HES and CEL associated with PDGFR gene re-arrangements were reported in 3 publications. The age of these patients ranged from 2 to 16 years and imatinib was given at dose 300 mg/m² daily or doses ranging from 200 to 400 mg daily. All patients achieved complete haematological response, complete cytogenetic response and/or complete molecular response.
Clinical studies in unresectable and/or metastatic GIST
One phase II, open-label, randomised, uncontrolled multinational study was conducted in patients with unresectable or metastatic malignant gastrointestinal stromal tumours (GIST). In this study 147 patients were enrolled and randomised to receive either 400 mg or 600 mg orally once daily for up to 36 months. These patients ranged in age from 18 to 83 years old and had a pathologic diagnosis of Kit-positive malignant GIST that was unresectable and/or metastatic. Immunohistochemistry was routinely performed with Kit antibody (A-4502, rabbit polyclonal antiserum, 1:100; DAKO Corporation, Carpinteria, CA) according to analysis by an avidin-biotin-peroxidase complex method after antigen retrieval.
The primary evidence of efficacy was based on objective response rates. Tumours were required to be measurable in at least one site of disease, and response characterisation based on Southwestern Oncology Group (SWOG) criteria. Results are provided in Table 6.
Table 6. Best tumour response in trial STIB2222 (GIST):
| All doses (n=147) | |
|---|---|
| 400 mg (n=73) | |
| 600 mg (n=74) | |
| Best response | n (%) |
| Complete response | 1(0.7) |
| Partial response | 98 (66.7) |
| Stable disease | 23 (15.6) |
| Progressive disease | 18 (12.2) |
| Not evaluable | 5 (3.4) |
| Unknown | 2 (1.4) |
There were no differences in response rates between the two dose groups. A significant number of patients who had stable disease at the time of the interim analysis achieved a partial response with longer treatment (median follow-up 31 months). Median time to response was 13 weeks (95% C.I. 12-23). Median time to treatment failure in responders was 122 weeks (95% C.I. 106-147), while in the overall study population it was 84 weeks (95% C.I. 71-109). The median overall survival has not been reached. The Kaplan-Meier estimate for survival after 36-month follow-up is 68%.
In two clinical studies (study B2222 and an intergroup study S0033) the daily dose of Glivec was escalated to 800 mg in patients progressing at the lower daily doses of 400 mg or 600 mg. The daily dose was escalated to 800 mg in a total of 103 patients; 6 patients achieved a partial response and 21 stabilisation of their disease after dose escalation for an overall clinical benefit of 26%. From the safety data available, escalating the dose to 800 mg daily in patients progressing at lower doses of 400 mg or 600 mg daily does not seem to affect the safety profile of Glivec.
Clinical studies in adjuvant GIST
In the adjuvant setting, Glivec was investigated in a multicentre, double-blind, long-term, placebo-controlled phase III study (Z9001) involving 773 patients. The ages of these patients ranged from 18 to 91 years. Patients were included who had a histological diagnosis of primary GIST expressing Kit protein by immunochemistry and a tumour size ≥3 cm in maximum dimension, with complete gross resection of primary GIST within 14-70 days prior to registration. After resection of primary GIST, patients were randomised to one of the two arms: Glivec at 400 mg/day or matching placebo for one year.
The primary endpoint of the study was recurrence-free survival (RFS), defined as the time from date of randomisation to the date of recurrence or death from any cause.
Glivec significantly prolonged RFS, with 75% of patients being recurrence-free at 38 months in the Glivec group vs. 20 months in the placebo group (95% CIs, [30 - non-estimable]; [14 - non-estimable], respectively); (hazard ratio = 0.398 [0.259-0.610], p<0.0001). At one year the overall RFS was significantly better for Glivec (97.7%) vs. placebo (82.3%), (p<0.0001). The risk of recurrence was thus reduced by approximately 89% as compared with placebo (hazard ratio = 0.113 [0.049-0.264]).
The risk of recurrence in patients after surgery of their primary GIST was retrospectively assessed based on the following prognostic factors: tumour size, mitotic index, tumour location. Mitotic index data were available for 556 of the 713 intention-to-treat (ITT) population. The results of subgroup analyses according to the United States National Institutes of Health (NIH) and the Armed Forces Institute of Pathology (AFIP) risk classifications are shown in Table 7. No benefit was observed in the low and very low risk groups. No overall survival benefit has been observed.
Table 7. Summary of Z9001 trial RFS analyses by NIH and AFIP risk classifications:
| Risk criteria | Risk Level | % of patients | No. of events/No. of patients | Overall hazard ratio (95%CI)* | RFS rates (%) | |
|---|---|---|---|---|---|---|
| 12 month | 24 month | |||||
| Glivec vs placebo | Glivec vs placebo | Glivec vs placebo | ||||
| NIH | Low | 29.5 | 0/86 vs. 2/90 | N.E. | 100 vs. 98.7 | 100 vs. 95.5 |
| Intermediate | 25.7 | 4/75 vs. 6/78 | 0.59 (0.17; 2.10) | 100 vs. 94.8 | 97.8 vs. 89.5 | |
| High | 44.8 | 21/140 vs. 51/127 | 0.29 (0.18; 0.49) | 94.8 vs. 64.0 | 80.7 vs. 46.6 | |
| AFIP | Very Low | 20.7 | 0/52 vs. 2/63 | N.E. | 100 vs. 98.1 | 100 vs. 93.0 |
| Low | 25.0 | 2/70 vs. 0/69 | N.E. | 100 vs. 100 | 97.8 vs. 100 | |
| Moderate | 24.6 | 2/70 vs. 11/67 | 0.16 (0.03; 0.70) | 97.9 vs. 90.8 | 97.9 vs. 73.3 | |
| High | 29.7 | 16/84 vs. 39/81 | 0.27 (0.15; 0.48) | 98.7 vs. 56.1 | 79.9 vs. 41.5 |
* Full follow-up period; NE – Not estimable
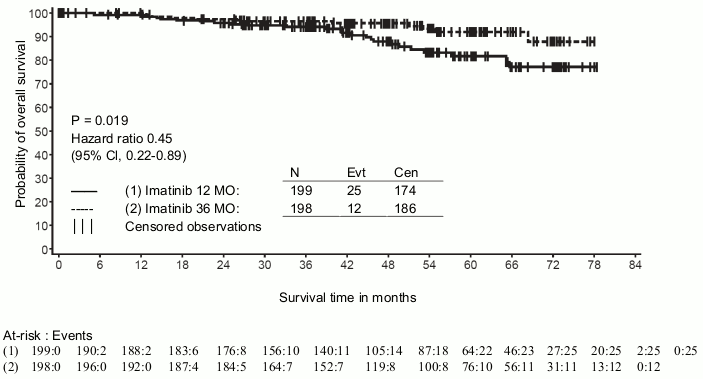
A second multicentre, open label phase III study (SSG XVIII/AIO) compared 400 mg/day Glivec 12 months treatment vs. 36 months treatment in patients after surgical resection of GIST and one of the following: tumour diameter >5 cm and mitotic count >5/50 high power fields (HPF); or tumour diameter >10 cm and any mitotic count or tumour of any size with mitotic count >10/50 HPF or tumours ruptured into the peritoneal cavity. There were a total of 397 patients consented and randomised to the study (199 patients on 12-month arm and 198 patients on 36-month arm), median age was 61 years (range 22 to 84 years). The median time of follow-up was 54 months (from date of randomisation to data cut-off), with a total of 83 months between the first patient randomised and the cut-off date.
The primary endpoint of the study was recurrence-free survival (RFS), defined as the time from date of randomisation to the date of recurrence or death from any cause.
Thirty-six (36) months of Glivec treatment significantly prolonged RFS compared to 12 months of Glivec treatment (with overall Hazard Ratio (HR) = 0.46 [0.32, 0.65], p<0.0001) (Table 8, Figure 1).
In addition, thirty-six (36) months of Glivec treatment significantly prolonged overall survival (OS) compared to 12 months of Glivec treatment (HR = 0.45 [0.22, 0.89], p=0.0187) (Table 8, Figure 2).
Longer duration of the treatment (>36 months) may delay the onset of further recurrences; however the impact of this finding on the overall survival remains unknown.
The total number of deaths were 25 for the 12-month treatment arm and 12 for the 36-month treatment arm.
Treatment with imatinib for 36 months was superior to treatment for 12 months in the ITT analysis, i.e. including the entire study population. In a planned subgroup analysis by mutation type, the HR for RFS for 36 months of treatment for patients with mutations of exon 11 was 0.35 [95% CI: 0.22, 0.56]. No conclusions can be drawn for other less common mutation subgroups due to the low number of observed events.
Table 8. 12-month and 36-month Glivec treatment (SSGXVIII/AIO Trial):
| 12-month treatment arm %(CI) | 36-month treatment arm %(CI) | |
|---|---|---|
| RFS | ||
| 12 months | 93.7 (89.2-96.4) | 95.9 (91.9-97.9) |
| 24 months | 75.4 (68.6-81.0) | 90.7 (85.6-94.0) |
| 36 months | 60.1 (52.5-66.9) | 86.6 (80.8-90.8) |
| 48 months | 52.3 (44.0-59.8) | 78.3 (70.8-84.1) |
| 60 months | 47.9 (39.0-56.3) | 65.6 (56.1-73.4) |
| Survival | ||
| 36 months | 94.0 (89.5-96.7) | 96.3 (92.4-98.2) |
| 48 months | 87.9 (81.1-92.3) | 95.6 (91.2-97.8) |
| 60 months | 81.7 (73.0-87.8) | 92.0 (85.3-95.7) |
Figure 1. Kaplan-Meier estimates for primary recurrence-free survival endpoint (ITT population):
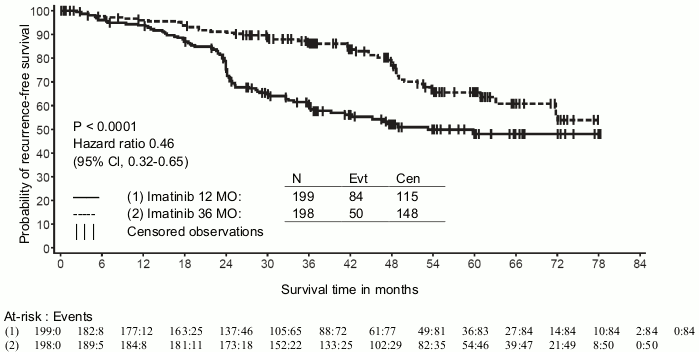
Figure 2. Kaplan-Meier estimates for overall survival (ITT population):
There are no controlled trials in paediatric patients with c-Kit positive GIST. Seventeen (17) patients with GIST (with or without Kit and PDGFR mutations) were reported in 7 publications. The age of these patients ranged from 8 to 18 years and imatinib was given in both adjuvant and metastatic settings at doses ranging from 300 to 800 mg daily. The majority of paediatric patients treated for GIST lacked data confirming c-kit or PDGFR mutations which may have led to mixed clinical outcomes.
Clinical studies in DFSP
One phase II, open label, multicentre clinical trial (study B2225) was conducted including 12 patients with DFSP treated with Glivec 800 mg daily. The age of the DFSP patients ranged from 23 to 75 years; DFSP was metastatic, locally recurrent following initial resective surgery and not considered amenable to further resective surgery at the time of study entry. The primary evidence of efficacy was based on objective response rates. Out of the 12 patients enrolled, 9 responded, one completely and 8 partially. Three of the partial responders were subsequently rendered disease free by surgery. The median duration of therapy in study B2225 was 6.2 months, with a maximum duration of 24.3 months. A further 6 DFSP patients treated with Glivec were reported in 5 published case reports, their ages ranging from 18 months to 49 years. The adult patients reported in the published literature were treated with either 400 mg (4 cases) or 800 mg (1 case) Glivec daily. Five (5) patients responded, 3 completely and 2 partially. The median duration of therapy in the published literature ranged between 4 weeks and more than 20 months. The translocation t(17:22)[(q22:q13)], or its gene product, was present in nearly all responders to Glivec treatment.
There are no controlled trials in paediatric patients with DFSP. Five (5) patients with DFSP and PDGFR gene re-arrangements were reported in 3 publications. The age of these patients ranged from newborn to 14 years and imatinib was given at dose 50 mg daily or doses ranging from 400 to 520 mg/m² daily. All patients achieved partial and/or complete response.5.2 Pharmacokinetic properties.
Pharmacokinetic properties
Pharmacokinetics of Glivec
The pharmacokinetics of Glivec have been evaluated over a dosage range of 25 to 1,000 mg. Plasma pharmacokinetic profiles were analysed on day 1 and on either day 7 or day 28, by which time plasma concentrations had reached steady state.
Absorption
Mean absolute bioavailability for imatinib is 98%. There was high between-patient variability in plasma imatinib AUC levels after an oral dose. When given with a high-fat meal, the rate of absorption of imatinib was minimally reduced (11% decrease in Cmax and prolongation of tmax by 1.5 h), with a small reduction in AUC (7.4%) compared to fasting conditions. The effect of prior gastrointestinal surgery on drug absorption has not been investigated.
Distribution
At clinically relevant concentrations of imatinib, binding to plasma proteins was approximately 95% on the basis of in vitro experiments, mostly to albumin and alpha-acid-glycoprotein, with little binding to lipoprotein.
Biotransformation
The main circulating metabolite in humans is the N-demethylated piperazine derivative, which shows similar in vitro potency to the parent. The plasma AUC for this metabolite was found to be only 16% of the AUC for imatinib. The plasma protein binding of the N-demethylated metabolite is similar to that of the parent compound.
Imatinib and the N-demethyl metabolite together accounted for about 65% of the circulating radioactivity (AUC(0-48h)). The remaining circulating radioactivity consisted of a number of minor metabolites.
The in vitro results showed that CYP3A4 was the major human P450 enzyme catalysing the biotransformation of imatinib. Of a panel of potential comedications (acetaminophen, aciclovir, allopurinol, amphotericin, cytarabine, erythromycin, fluconazole, hydroxyurea, norfloxacin, penicillin V) only erythromycin (IC50 50 μΜ) and fluconazole (IC50 118 μΜ) showed inhibition of imatinib metabolism which could have clinical relevance.
Imatinib was shown in vitro to be a competitive inhibitor of marker substrates for CYP2C9, CYP2D6 and CYP3A4/5. Ki values in human liver microsomes were 27, 7.5 and 7.9 μmol/l, respectively. Maximal plasma concentrations of imatinib in patients are 2-4 μmol/l, consequently an inhibition of CYP2D6 and/or CYP3A4/5-mediated metabolism of co-administered drugs is possible. Imatinib did not interfere with the biotransformation of 5-fluorouracil, but it inhibited paclitaxel metabolism as a result of competitive inhibition of CYP2C8 (Ki = 34.7 μΜ). This Ki value is far higher than the expected plasma levels of imatinib in patients, consequently no interaction is expected upon co-administration of either 5-fluorouracil or paclitaxel and imatinib.
Elimination
Based on the recovery of compound(s) after an oral 14C-labelled dose of imatinib, approximately 81% of the dose was recovered within 7 days in faeces (68% of dose) and urine (13% of dose). Unchanged imatinib accounted for 25% of the dose (5% urine, 20% faeces), the remainder being metabolites.
Plasma pharmacokinetics
Following oral administration in healthy volunteers, the ty2 was approximately 18 h, suggesting that once-daily dosing is appropriate. The increase in mean AUC with increasing dose was linear and dose proportional in the range of 25-1,000 mg imatinib after oral administration. There was no change in the kinetics of imatinib on repeated dosing, and accumulation was 1.5-2.5-fold at steady state when dosed once daily.
Pharmacokinetics in GIST patients
In patients with GIST steady-state exposure was 1.5-fold higher than that observed for CML patients for the same dosage (400 mg daily). Based on preliminary population pharmacokinetic analysis in GIST patients, there were three variables (albumin, WBC and bilirubin) found to have a statistically significant relationship with imatinib pharmacokinetics. Decreased values of albumin caused a reduced clearance (CL/f); and higher levels of WBC led to a reduction of CL/f. However, these associations are not sufficiently pronounced to warrant dose adjustment. In this patient population, the presence of hepatic metastases could potentially lead to hepatic insufficiency and reduced metabolism.
Population pharmacokinetics
Based on population pharmacokinetic analysis in CML patients, there was a small effect of age on the volume of distribution (12% increase in patients >65 years old). This change is not thought to be clinically significant. The effect of bodyweight on the clearance of imatinib is such that for a patient weighing 50 kg the mean clearance is expected to be 8.5 l/h, while for a patient weighing 100 kg the clearance will rise to 11.8 l/h. These changes are not considered sufficient to warrant dose adjustment based on kg bodyweight. There is no effect of gender on the kinetics of imatinib.
Pharmacokinetics in children
As in adult patients, imatinib was rapidly absorbed after oral administration in paediatric patients in both phase I and phase II studies. Dosing in children at 260 and 340 mg/m²/day achieved the same exposure, respectively, as doses of 400 mg and 600 mg in adult patients. The comparison of AUC(0-24) on day 8 and day 1 at the 340 mg/m²/day dose level revealed a 1.7-fold drug accumulation after repeated once-daily dosing.
Based on pooled population pharmacokinetic analysis in paediatric patients with haematological disorders (CML, Ph+ALL, or other haematological disorders treated with imatinib), clearance of imatinib increases with increasing body surface area (BSA). After correcting for the BSA effect, other demographics such as age, body weight and body mass index did not have clinically significant effects on the exposure of imatinib. The analysis confirmed that exposure of imatinib in paediatric patients receiving 260 mg/m² once daily (not exceeding 400 mg once daily) or 340 mg/m² once daily (not exceeding 600 mg once daily) were similar to those in adult patients who received imatinib 400 mg or 600 mg once daily.
Organ function impairment
Imatinib and its metabolites are not excreted via the kidney to a significant extent. Patients with mild and moderate impairment of renal function appear to have a higher plasma exposure than patients with normal renal function. The increase is approximately 1.5- to 2-fold, corresponding to a 1.5-fold elevation of plasma AGP, to which imatinib binds strongly. The free drug clearance of imatinib is probably similar between patients with renal impairment and those with normal renal function, since renal excretion represents only a minor elimination pathway for imatinib (see sections 4.2 and 4.4).
Although the results of pharmacokinetic analysis showed that there is considerable inter-subject variation, the mean exposure to imatinib did not increase in patients with varying degrees of liver dysfunction as compared to patients with normal liver function (see sections 4.2, 4.4 and 4.8).
Preclinical safety data
The preclinical safety profile of imatinib was assessed in rats, dogs, monkeys and rabbits.
Multiple dose toxicity studies revealed mild to moderate haematological changes in rats, dogs and monkeys, accompanied by bone marrow changes in rats and dogs.
The liver was a target organ in rats and dogs. Mild to moderate increases in transaminases and slight decreases in cholesterol, triglycerides, total protein and albumin levels were observed in both species. No histopathological changes were seen in rat liver. Severe liver toxicity was observed in dogs treated for 2 weeks, with elevated liver enzymes, hepatocellular necrosis, bile duct necrosis, and bile duct hyperplasia.
Renal toxicity was observed in monkeys treated for 2 weeks, with focal mineralisation and dilation of the renal tubules and tubular nephrosis. Increased blood urea nitrogen (BUN) and creatinine were observed in several of these animals. In rats, hyperplasia of the transitional epithelium in the renal papilla and in the urinary bladder was observed at doses ≥6 mg/kg in the 13-week study, without changes in serum or urinary parameters. An increased rate of opportunistic infections was observed with chronic imatinib treatment.
In a 39-week monkey study, no NOAEL (no observed adverse effect level) was established at the lowest dose of 15 mg/kg, approximately one-third the maximum human dose of 800 mg based on body surface. Treatment resulted in worsening of normally suppressed malarial infections in these animals.
Imatinib was not considered genotoxic when tested in an in vitro bacterial cell assay (Ames test), an in vitro mammalian cell assay (mouse lymphoma) and an in vivo rat micronucleus test. Positive genotoxic effects were obtained for imatinib in an in vitro mammalian cell assay (Chinese hamster ovary) for clastogenicity (chromosome aberration) in the presence of metabolic activation. Two intermediates of the manufacturing process, which are also present in the final product, are positive for mutagenesis in the Ames assay. One of these intermediates was also positive in the mouse lymphoma assay.
In a study of fertility, in male rats dosed for 70 days prior to mating, testicular and epididymal weights and percent motile sperm were decreased at 60 mg/kg, approximately equal to the maximum clinical dose of 800 mg/day, based on body surface area. This was not seen at doses ≤20 mg/kg. A slight to moderate reduction in spermatogenesis was also observed in the dog at oral doses ≥30 mg/kg. When female rats were dosed 14 days prior to mating and through to gestational day 6, there was no effect on mating or on number of pregnant females. At a dose of 60 mg/kg, female rats had significant post-implantation foetal loss and a reduced number of live foetuses. This was not seen at doses ≤20 mg/kg.
In an oral pre- and postnatal development study in rats, red vaginal discharge was noted in the 45 mg/kg/day group on either day 14 or day 15 of gestation. At the same dose, the number of stillborn pups as well as those dying between postpartum days 0 and 4 was increased. In the F1 offspring, at the same dose level, mean body weights were reduced from birth until terminal sacrifice and the number of litters achieving criterion for preputial separation was slightly decreased. F1 fertility was not affected, while an increased number of resorptions and a decreased number of viable foetuses was noted at 45 mg/kg/day. The no observed effect level (NOEL) for both the maternal animals and the F1 generation was 15 mg/kg/day (one quarter of the maximum human dose of 800 mg).
Imatinib was teratogenic in rats when administered during organogenesis at doses ≥100 mg/kg, approximately equal to the maximum clinical dose of 800 mg/day, based on body surface area. Teratogenic effects included exencephaly or encephalocele, absent/reduced frontal and absent parietal bones. These effects were not seen at doses ≤30 mg/kg.
No new target organs were identified in the rat juvenile development toxicology study (day 10 to 70 postpartum) with respect to the known target organs in adult rats. In the juvenile toxicology study, effects upon growth, delay in vaginal opening and preputial separation were observed at approximately 0.3 to 2 times the average paediatric exposure at the highest recommended dose of 340 mg/m². In addition, mortality was observed in juvenile animals (around weaning phase) at approximately 2 times the average paediatric exposure at the highest recommended dose of 340 mg/m².
In the 2-year rat carcinogenicity study administration of imatinib at 15, 30 and 60 mg/kg/day resulted in a statistically significant reduction in the longevity of males at 60 mg/kg/day and females at ≥30 mg/kg/day. Histopathological examination of decedents revealed cardiomyopathy (both sexes), chronic progressive nephropathy (females) and preputial gland papilloma as principal causes of death or reasons for sacrifice. Target organs for neoplastic changes were the kidneys, urinary bladder, urethra, preputial and clitoral gland, small intestine, parathyroid glands, adrenal glands and non-glandular stomach.
Papilloma/carcinoma of the preputial/clitoral gland were noted from 30 mg/kg/day onwards, representing approximately 0.5 or 0.3 times the human daily exposure (based on AUC) at 400 mg/day or 800 mg/day, respectively, and 0.4 times the daily exposure in children (based on AUC) at 340 mg/m²/day. The no observed effect level (NOEL) was 15 mg/kg/day. The renal adenoma/carcinoma, the urinary bladder and urethra papilloma, the small intestine adenocarcinomas, the parathyroid glands adenomas, the benign and malignant medullary tumours of the adrenal glands and the non-glandular stomach papillomas/carcinomas were noted at 60 mg/kg/day, representing approximately 1.7 or 1 times the human daily exposure (based on AUC) at 400 mg/day or 800 mg/day, respectively, and 1.2 times the daily exposure in children (based on AUC) at 340 mg/m²/day. The no observed effect level (NOEL) was 30 mg/kg/day.
The mechanism and relevance of these findings in the rat carcinogenicity study for humans are not yet clarified.
Non-neoplastic lesions not identified in earlier preclinical studies were the cardiovascular system, pancreas, endocrine organs and teeth. The most important changes included cardiac hypertrophy and dilatation, leading to signs of cardiac insufficiency in some animals.
The active substance imatinib demonstrates an environmental risk for sediment organisms.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.