KAYFANDA Hard capsule Ref.[115085] Active ingredients: Odevixibat
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Ipsen Pharma, 65 quai Georges Gorse, 92100 Boulogne-Billancourt, France
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Bile and liver therapy, other drugs for bile therapy
ATC code: A05AX05
Mechanism of action
Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT).
Odevixibat acts locally in the distal ileum to decrease the reuptake of bile acids and increase the clearance of bile acids through the colon, reducing the concentration of bile acids in the serum. The extent of reduction of serum bile acids does not correlate with systemic pharmacokinetics (PK).
Clinical efficacy
The efficacy of odevixibat in patients with ALGS was evaluated in two phase 3 trials. Study A4250- 012 (ASSERT) was a 24-week, randomised, double-blind, placebo-controlled trial conducted in 52 patients with a confirmed diagnosis of ALGS. Patients were randomised 2:1 to 120 mcg/kg/day odevixibat or placebo and stratified by age at randomisation (<10 years and ≥10 to <18 years). Patients whose ALT was >10 × upper limit of normal (ULN) or total bilirubin >15 × ULN at screening were excluded in the ASSERT trial.
The primary endpoint in ASSERT was change in scratching severity score from baseline to month 6 (weeks 21 to 24) based on the worst scratching score using an observer-reported outcome (ObsRO) instrument. Scratching was assessed once in the morning and once in the evening using a 5-point scale (0-4).
Change in serum bile acid levels from baseline to the average of weeks 20 and 24 was the key secondary endpoint. Additional secondary endpoints included change from baseline to end of treatment in sleep parameters (assessed using a 5-point scale (0-4)), total cholesterol concentration and clinician assessment of xanthomas.
Median age (range) of the patients in ASSERT was 5.45 (0.5 to 15.5) years; 51.9% were male and 82.7% were white. 92.3% of patients had the Jagged canonical NOTCH ligand 1 (JAG1) mutation and 7.7% had the NOTCH2 mutation. At baseline, 98.1% of patients were treated with concomitant anti-pruritic medications, including UDCA (88.5%). Overall, 51 (98.1%) of the 52 patients had moderate hepatic impairment and 1 (1.9%) (placebo group) had severe hepatic impairment based on the Child- Pugh C classification. Baseline mean (standard deviation [SD)] estimated glomerular filtration rate (eGFR) was 158.65 (51.437) mL/min/1.73 m². Baseline mean (SD) ALT, AST, and total bilirubin were 173.7 (84.48) U/L, 167.0 (83.22) U/L, and 55.14 (47.911) μmol/L, respectively. Baseline mean (SD) scratching score (range: 0-4) and serum bile acids levels were similar in odevixibat-treated patients (2.80 [0.520] and 237.4 [114.88] μmol/L, respectively) and placebo-treated patients (3.01 [0.636] and 246.1 [120.53] μmol/L, respectively).
Table 4 presents the results of the change from baseline in average scratching score based on the ObsRO assessments to month 6 (weeks 21 to 24) and results of the change from baseline in serum bile acids to the average of weeks 20 and 24.
Table 4. Comparison of key efficacy results for odevixibat vs. placebo over the 24-week treatment period (ASSERT):
| Placebo (N=17) | Odevixibat 120 mcg/kg/day (N=35) | |
|---|---|---|
| Change from baseline in average scratching scorea to month 6 (Weeks 21 to 24) of treatment | ||
| LS Mean (95% CI)b | -0.80 (-1.27, -0.33) | -1.69 (-2.04, -1.34) |
| LS Mean difference vs. placebo (95% CI)b Two-sided p-valueb | -0.88 (-1.44, -0.33) 0.0025 | |
| Change from baseline in serum bile acid concentration (μmol/L) to the average of weeks 20 and 24 of treatment | ||
| LS Mean (95% CI)b | 22.39 (-34.75, 79.52) | -90.35 (-1.33, -47.56) |
| LS Mean difference vs. placebo (95% CI)b Two-sided p-valueb | -112.74 (-178.78, -46.69) 0.0012 | |
CI: confidence interval; LS Mean = Least Squares Means
a Based on the ObsRO instrument which is a validated 0-4 scale completed by caregivers (0=none to 4=very severe), where changes ≥1.0 have been shown to be clinically meaningful.
b The analyses are based on mixed-model effect repeated measures (MMRM) with baseline scratching score or baseline serum bile acid concentration (as applicable for the endpoint) as a covariate, and baseline age stratification (<10, ≥10 years), baseline direct bilirubin (scratching score only), treatment group, time (months/visits), and treatment-by-time interaction as fixed effects.
Figures 1 and 2 display graphically the mean changes standard error (SE) from baseline of patients' average scratching scores in each treatment group for each week and patients' serum bile acid levels in each treatment group for each month, respectively.
Figure 1. Mean (± SE) change from baseline in pruritus (scratching) severity score over time (ASSERT):
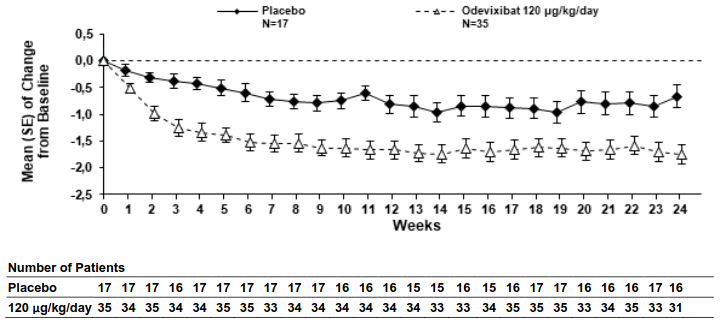
Figure 2. Mean (± SE) change from baseline in serum bile acid concentration (μmol/L) over time (ASSERT):
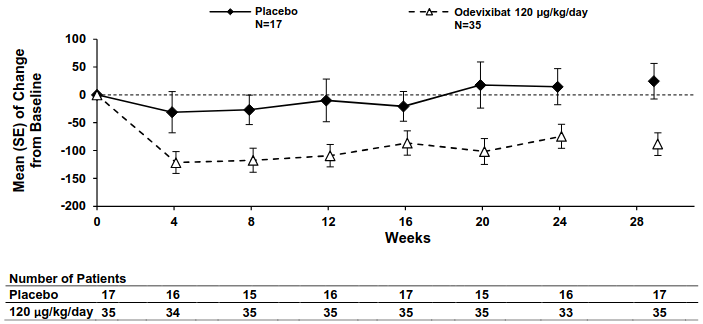
Consistent with the results for improvement in pruritus (scratching) severity, odevixibat led to improvements in multiple sleep parameters. Figure 3 displays graphically the mean changes (SE) from baseline for improvement in two of the sleep parameters by treatment group for each month, including percentage of days with help falling asleep and daytime tiredness score. Similar results were observed over time for percentage of days the child required soothing to go to sleep and the percentage of days the child slept with the caregiver.
Figure 3. Mean (±SE) change from baseline in sleep parameters over time (ASSERT):
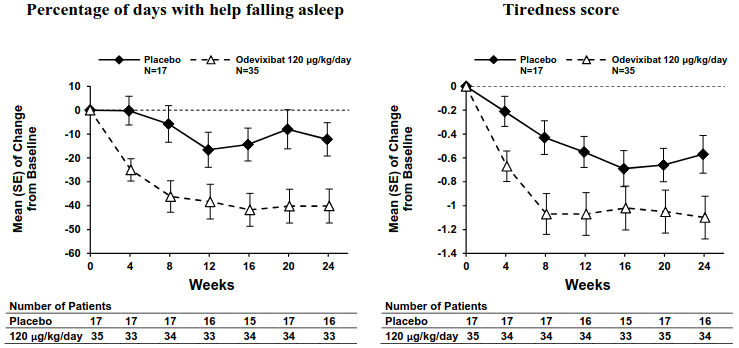
Study A4250-015 (ASSERT-EXT) is a 72-week, single-arm, open-label extension trial in patients with ALGS who completed ASSERT. Continued treatment with odevixibat 120 mcg/kg/day in ASSERT-EXT led to further improvements in pruritus score and sleep parameters and reduction in serum bile acids, all of which were sustained through 72 weeks of treatment. Improvements from baseline in these endpoints were observed in patients who received placebo in ASSERT. In ASSERT-EXT, 42 of 50 patients had received odevixibat treatment for more than 72 weeks; 6 patients had discontinued prior to 72 weeks of treatment with odevixibat.
Exceptional circumstances
This medicinal product has been authorised under ‘Exceptional Circumstances’. This means that due to the rarity of the disease it has not been possible to obtain complete information on this medicinal product. The European Medicines Agency will review any new information which may become available every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Absorption
Odevixibat is minimally absorbed following oral administration; absolute bioavailability data in humans are not available, and estimated relative bioavailability is <1%. Peak odevixibat plasma concentration (Cmax) is reached within 1 to 5 hours. Observed exposures in paediatric patients (age between 0.756 and 17.1 years; body weight from 5.6 to 58 kg) are limited to trough values; for the 120 mcg/kg/day dose the trough values were below the limit of detection for 40% of the samples in ALGS patients. The mean Cmax value in a paediatric ALGS patient population for the 120 mcg/kg/day dose is 1.13 ng/mL and the mean AUC value was 13.2 ng × h/mL. No accumulation of odevixibat was observed following once-daily dosing.
Effect of food
Systemic exposure of odevixibat does not predict efficacy. Therefore, no dose adjustment for food effects is considered necessary. Concomitant administration of a high-fat meal (800 – 1 000 calories with approximately 50% of total caloric content of the meal from fat) resulted in decreases of approximately 72% and 62% in Cmax and AUC0-24, respectively, compared to administration under fasted conditions. When odevixibat was sprinkled on apple sauce, decreases of approximately 39% and 36% in Cmax and AUC0-24, respectively, were observed compared to administration under fasted conditions. Taking into account the lack of PK/pharmacodynamic (PD) relationship and need for sprinkling the odevixibat capsule contents on food for younger children, odevixibat can be administered with food.
Distribution
Odevixibat is more than 99% bound to human plasma proteins. The mean volume of distribution (V/F) in ALGS patients is predicted to be 1160 L. The geometric mean body weight adjusted V/F for ALGS is 57.9 L/kg.
Biotransformation
Odevixibat is minimally metabolised in humans.
Elimination
Following administration of a single oral dose of 3 000 mcg of radiolabeled odevixibat in healthy adults, the average percent recovery of the administered dose was 82.9% in faeces; less than 0.002% was recovered in the urine. More than 97% of faecal radioactivity was determined to be unchanged odevixibat.
The mean apparent clearance (CL/F) in ALGS patients is predicted to be 212 L/h, and the mean half-life is approximately 4.75 hours. The geometric mean body weight adjusted CL/F for ALGS is 10.5 L/h/kg.
Linearity/non-linearity
The Cmax and AUC0-t increase with increasing doses in a dose-proportional manner; however due to the high interindividual variability of approximately 40%, it is not possible to estimate the dose proportionality accurately.
Pharmacokinetic/pharmacodynamic relationship(s)
Consistent with the mechanism and site of action of odevixibat in the gastrointestinal tract no relationship between systemic exposure and clinical effects is observed. Also, no dose-response relationship could be established for the investigated dose range 10-200 mcg/kg/day and the PD parameters 7α-hydroxy-4 cholesten-3 one (C4) and fibroblast growth factor 19 (FGF19).
Special populations
No clinically significant differences in the PK of odevixibat were observed based on age, sex, or race.
Hepatic impairment
All patients with ALGS presented with some degree of hepatic impairment because of the disease. Based on population PK analysis, patients with mild hepatic impairment (Child-Pugh A) exhibited comparable pharmacokinetics compared to other subjects with normal hepatic function. Patients with moderate hepatic impairment (Child-Pugh B) presented a 77.0% lower CL/F relative to patients with mild hepatic impairment or no hepatic impairment and exposure of odevixibat was 4- to 9-fold higher in patients with moderate hepatic impairment. The plasma AUC at 120 mcg/kg/day in patients with Child-Pugh A ranged from 1.52 to 10.4 ng × h/mL and in patients with Child-Pugh B was between 5.50 to 74.5 ng × h/mL. Although, CL/F values were lower and V/F values were larger in patients with Child Pugh B compared to other subjects, no accumulation of odevixibat was observed and the safety profile was comparable between the patient groups. No data are available for patients with severe hepatic impairment (Child-Pugh C).
Renal impairment
There are no clinical data in patients with renal impairment, but the impact of renal impairment is expected to be small due to low systemic exposure and odevixibat not being excreted in urine.
In vitro studies
In in vitro studies, odevixibat did not inhibit CYPs 1A2, 2B6, 2C8, 2C9, 2C19 or 2D6 at clinically relevant concentrations, but was shown to be an inhibitor of CYP3A4/5.
Odevixibat does not inhibit the transporters P-gp, breast cancer resistance protein (BCRP), organic anion transporters (OATP1B1, OATP1B3, OAT1, OAT3), organic cation transporter (OCT2), multidrug and toxin extrusion transporters (MATE1 or MATE2-K).
Odevixibat is a substrate of gastrointestinal efflux transporter P-gp, but not of BCRP.
5.3. Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows:
Reproductive and developmental toxicity
In pregnant New Zealand White rabbits, early delivery/abortion was observed in two rabbits receiving odevixibat during the period of foetal organogenesis at an exposure multiple of ≥1.6 of the anticipated clinical exposure (based on total plasma odevixibat AUC0-24). Reductions in maternal body weight and food consumption were noted in all dose groups (transient at the exposure multiple 0.5 of the anticipated dose).
Starting from the exposure multiple of 0.5 of the clinical human exposure (based on total plasma odevixibat AUC0-24), 7 foetuses (1.3% of all foetuses from odevixibat exposed does) in all dose groups were found to have cardiovascular defects (i.e. ventricular diverticulum, small ventricle and dilated aortic arch). No such malformations were observed when odevixibat was administered to pregnant rats. Because of the findings in rabbits, an effect of odevixibat on cardiovascular development cannot be excluded.
Odevixibat had no effect on the reproductive performance, fertility, embryo-foetal development, or prenatal/postnatal development studies in rats at the exposure multiple of 133 of the anticipated clinical exposure (based on total plasma odevixibat AUC0-24), including juveniles (exposure multiple of 63 of the anticipated human exposure).
There is insufficient information on the excretion of odevixibat in animal milk. The presence of odevixibat in breast milk was not measured in animal studies. Exposure was demonstrated in the pups of lactating dams in the pre- and post-natal developmental toxicity study with rats (3.2-52.1% of the odevixibat plasma concentration of the lactating dams). It is therefore possible that odevixibat is present in breast milk.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.