Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: STADA Arzneimittel AG, Stadastrasse 2–18, 61118 Bad Vilbel, Germany
Pharmacotherapeutic group: Immunosuppressants, Tumour necrosis factor alpha (TNF-α) inhibitors
ATC code: L04AB04
Adalimumab binds specifically to TNF and neutralises the biological function of TNF by blocking its interaction with the p55 and p75 cell surface TNF receptors.
Adalimumab also modulates biological responses that are induced or regulated by TNF, including changes in the levels of adhesion molecules responsible for leukocyte migration (ELAM-1, VCAM-1, and ICAM-1 with an IC50 of 0.1-0.2 nM).
After treatment with adalimumab, a rapid decrease in levels of acute phase reactants of inflammation (C-reactive protein (CRP) and erythrocyte sedimentation rate (ESR)) and serum cytokines (IL-6) was observed, compared to baseline in patients with rheumatoid arthritis. Serum levels of matrix metalloproteinases (MMP-1 and MMP-3) that produce tissue remodelling responsible for cartilage destruction were also decreased after adalimumab administration. Patients treated with adalimumab usually experienced improvement in haematological signs of chronic inflammation.
A rapid decrease in CRP levels was also observed in patients with polyarticular juvenile idiopathic arthritis, Crohn’s disease, ulcerative colitis and HS after treatment with adalimumab. In patients with Crohn’s disease, a reduction of the number of cells expressing inflammatory markers in the colon including a significant reduction of expression of TNFα was seen. Endoscopic studies in intestinal mucosa have shown evidence of mucosal healing in adalimumab treated patients.
Adalimumab was evaluated in over 3,000 patients in all rheumatoid arthritis clinical trials. The efficacy and safety of adalimumab were assessed in five randomised, double-blind and well-controlled studies. Some patients were treated for up to 120 months duration. Injection site pain of adalimumab 40 mg/0.4 ml was assessed in two randomised, active control, single-blind, two-period crossover studies.
RA study I evaluated 271 patients with moderately to severely active rheumatoid arthritis who were ≥18 years old, had failed therapy with at least one disease-modifying, anti-rheumatic drug and had insufficient efficacy with methotrexate at doses of 12.5 to 25 mg (10 mg if methotrexate-intolerant) every week and whose methotrexate dose remained constant at 10 to 25 mg every week. Doses of 20, 40 or 80 mg of adalimumab or placebo were given every other week for 24 weeks.
RA study II evaluated 544 patients with moderately to severely active rheumatoid arthritis who were ≥18 years old and had failed therapy with at least one disease-modifying, anti-rheumatic drugs. Doses of 20 mg or 40 mg of adalimumab were given by subcutaneous injection every other week with placebo on alternative weeks or every week for 26 weeks; placebo was given every week for the same duration. No other disease-modifying anti-rheumatic drugs were allowed.
RA study III evaluated 619 patients with moderately to severely active rheumatoid arthritis who were ≥18 years old, and who had an ineffective response to methotrexate at doses of 12.5 to 25 mg or have been intolerant to 10 mg of methotrexate every week. There were three groups in this study. The first received placebo injections every week for 52 weeks. The second received 20 mg of adalimumab every week for 52 weeks. The third group received 40 mg of adalimumab every other week with placebo injections on alternate weeks. Upon completion of the first 52 weeks, 457 patients enroled in an open-label extension phase in which 40 mg of adalimumab/MTX was administered every other week up to 10 years.
RA study IV primarily assessed safety in 636 patients with moderately to severely active rheumatoid arthritis who were ≥18 years old. Patients were permitted to be either disease-modifying, antirheumatic drug-naïve or to remain on their pre-existing rheumatologic therapy provided that therapy was stable for a minimum of 28 days. These therapies include methotrexate, leflunomide, hydroxychloroquine, sulfasalazine and/or gold salts. Patients were randomised to 40 mg of adalimumab or placebo every other week for 24 weeks.
RA study V evaluated 799 methotrexate-naïve, adult patients with moderate to severely active early rheumatoid arthritis (mean disease duration less than 9 months). This study evaluated the efficacy of adalimumab 40 mg every other week/methotrexate combination therapy, adalimumab 40 mg every other week monotherapy and methotrexate monotherapy in reducing the signs and symptoms and rate of progression of joint damage in rheumatoid arthritis for 104 weeks. Upon completion of the first 104 weeks, 497 patients enroled in an open-label extension phase in which 40 mg of adalimumab was administered every other week up to 10 years.
RA studies VI and VII each evaluated 60 patients with moderately to severely active rheumatoid arthritis who were ≥ 18 years old. Enroled patients were either current users of adalimumab 40 mg/0.8 ml and rated their average injection site pain as at least 3 cm (on a 0-10 cm VAS) or were biologic-naïve subjects who were starting adalimumab 40 mg/0.8 ml. Patients were randomised to receive a single dose of adalimumab 40 mg/0.8 ml or adalimumab 40 mg/0.4 ml, followed by a single injection of the opposite treatment at their next dose.
The primary end point in RA studies I, II and III and the secondary endpoint in RA study IV was the percent of patients who achieved an ACR 20 response at Week 24 or 26. The primary endpoint in RA study V was the percent of patients who achieved an ACR 50 response at Week 52. RA studies III and V had an additional primary endpoint at 52 weeks of retardation of disease progression (as detected by X-ray results). RA study III also had a primary endpoint of changes in quality of life. The primary endpoint in RA studies VI and VII was injection site pain immediately after injection as measured by a 0-10 cm VAS.
The percent of adalimumab-treated patients achieving ACR 20, 50 and 70 responses was consistent across RA studies I, II and III. The results for the 40 mg every other week dose are summarised in Table 5.
Table 5. ACR responses in placebo-controlled trials (percent of patients):
| Response | RA Study Ia** | RA Study IIa** | RA Study IIIa** | |||
|---|---|---|---|---|---|---|
| Placebo/ MTXc n=60 | Adalimumabb/ MTXc n=63 | Placebo n=110 | Adalimumabb n=113 | Placebo/ MTXc n=200 | Adalimumabb/ MTXc n=207 | |
| ACR 20 | ||||||
| 6 months | 13.3% | 65.1% | 19.1% | 46.0% | 29.5% | 63.3% |
| 12 months | NA | NA | NA | NA | 24.0% | 58.9% |
| ACR 50 | ||||||
| 6 months | 6.7% | 52.4% | 8.2% | 22.1% | 9.5% | 39.1% |
| 12 months | NA | NA | NA | NA | 9.5% | 41.5% |
| ACR 70 | ||||||
| 6 months | 3.3% | 23.8% | 1.8% | 12.4% | 2.5% | 20.8% |
| 12 months | NA | NA | NA | NA | 4.5% | 23.2% |
a RA study I at 24 weeks, RA study II at 26 weeks, and RA study III at 24 and 52 weeks
b 40 mg adalimumab administered every other week
c MTX = methotrexate
** p<0.01, adalimumab versus placebo
In RA studies I-IV, all individual components of the ACR response criteria (number of tender and swollen joints, physician and patient assessment of disease activity and pain, disability index (HAQ) scores and CRP (mg/dl) values) improved at 24 or 26 weeks compared to placebo. In RA study III, these improvements were maintained throughout 52 weeks.
In the open-label extension for RA study III, most patients who were ACR responders maintained response when followed for up to 10 years. Of 207 patients who were randomised to adalimumab 40 mg every other week, 114 patients continued on adalimumab 40 mg every other week for 5 years. Among those, 86 patients (75.4%) had ACR 20 responses; 72 patients (63.2%) had ACR 50 responses; and 41 patients (36%) had ACR 70 responses. Of 207 patients, 81 patients continued on adalimumab 40 mg every other week for 10 years. Among those, 64 patients (79.0%) had ACR 20 responses; 56 patients (69.1%) had ACR 50 responses; and 43 patients (53.1%) had ACR 70 responses.
In RA study IV, the ACR 20 response of patients treated with adalimumab plus standard of care was statistically significantly better than patients treated with placebo plus standard of care (p<0.001).
In RA studies I-IV, adalimumab-treated patients achieved statistically significant ACR 20 and 50 responses compared to placebo as early as one to two weeks after initiation of treatment.
In RA study V with early rheumatoid arthritis patients who were methotrexate naïve, combination therapy with adalimumab and methotrexate led to faster and significantly greater ACR responses than methotrexate monotherapy and adalimumab monotherapy at Week 52 and responses were sustained at Week 104 (see Table 6).
Table 6. ACR responses in RA study V (percent of patients):
| Response | MTX n=257 | Adalimumab n=274 | Adalimumab/MTX n=268 | p-valuea | p-valueb | p-valuec |
|---|---|---|---|---|---|---|
| ACR 20 | ||||||
| Week 52 | 62.6% | 54.4% | 72.8% | 0.013 | <0.001 | 0.043 |
| Week 104 | 56.0% | 49.3% | 69.4% | 0.002 | <0.001 | 0.140 |
| ACR 50 | ||||||
| Week 52 | 45.9% | 41.2% | 61.6% | <0.001 | <0.001 | 0.317 |
| Week 104 | 42.8% | 36.9% | 59.0% | <0.001 | <0.001 | 0.162 |
| ACR 70 | ||||||
| Week 52 | 27.2% | 25.9% | 45.5% | <0.001 | <0.001 | 0.656 |
| Week 104 | 28.4% | 28.1% | 46.6% | <0.001 | <0.001 | 0.864 |
a p-value is from the pairwise comparison of methotrexate monotherapy and adalimumab/methotrexate combination therapy using the Mann-Whitney U test.
b p-value is from the pairwise comparison of adalimumab monotherapy and adalimumab/methotrexate combination therapy using the Mann-Whitney U test
c p-value is from the pairwise comparison of adalimumab monotherapy and methotrexate monotherapy using the Mann-Whitney U test
In the open-label extension for RA study V, ACR response rates were maintained when followed for up to 10 years. Of 542 patients who were randomised to adalimumab 40 mg every other week, 170 patients continued on adalimumab 40 mg every other week for 10 years. Among those, 154 patients (90.6%) had ACR 20 responses; 127 patients (74.7%) had ACR 50 responses; and 102 patients (60.0%) had ACR 70 responses.
At Week 52, 42.9% of patients who received adalimumab/methotrexate combination therapy achieved clinical remission (DAS28 (CRP) <2.6) compared to 20.6% of patients receiving methotrexate monotherapy and 23.4% of patients receiving adalimumab monotherapy. Adalimumab/methotrexate combination therapy was clinically and statistically superior to methotrexate (p<0.001) and adalimumab monotherapy (p<0.001) in achieving a low disease state in patients with recently diagnosed moderate to severe rheumatoid arthritis. The response for the two monotherapy arms was similar (p=0.447). Of 342 subjects originally randomised to adalimumab monotherapy or adalimumab/methotrexate combination therapy who entered the open-label extension study, 171 subjects completed 10 years of adalimumab treatment. Among those, 109 subjects (63.7%) were reported to be in remission at 10 years.
In RA study III, where adalimumab-treated patients had a mean duration of rheumatoid arthritis of approximately 11 years, structural joint damage was assessed radiographically and expressed as change in modified Total Sharp Score (TSS) and its components, the erosion score and joint space narrowing score. Adalimumab/methotrexate patients demonstrated significantly less radiographic progression than patients receiving methotrexate alone at 6 and 12 months (see Table 7).
In the open-label extension of RA Study III, the reduction in rate of progression of structural damage is maintained for 8 and 10 years in a subset of patients. At 8 years, 81 of 207 patients originally treated with 40 mg adalimumab every other week were evaluated radiographically. Among those, 48 patients showed no progression of structural damage defined by a change from baseline in the mTSS of 0.5 or less. At 10 years, 79 of 207 patients originally treated with 40 mg adalimumab every other week were evaluated radiographically. Among those, 40 patients showed no progression of structural damage defined by a change from baseline in the mTSS of 0.5 or less.
Table 7. Radiographic mean changes over 12 months in RA study III:
| Placebo/ MTXa | Adalimumab/MTX 40 mg every other week | Placebo/MTX- Adalimumab/MTX (95% Confidence Intervalb) | p-value | |
|---|---|---|---|---|
| Total Sharp Score | 2.7 | 0.1 | 2.6 (1.4, 3.8) | <0.001c |
| Erosion score | 1.6 | 0.0 | 1.6 (0.9, 2.2) | <0.001 |
| JSNd score | 1.0 | 0.1 | 0.9 (0.3, 1.4) | 0.002 |
a methotrexate
b 95% confidence intervals for the differences in change scores between methotrexate and adalimumab
c Based on rank analysis
d Joint Space Narrowing
In RA study V, structural joint damage was assessed radiographically and expressed as change in modified Total Sharp Score (see Table 8).
Table 8. Radiographic mean changes at Week 52 in RA study V:
| MTX n=257 (95% confidence interval) | Adalimumab n=274 (95% confidence interval) | Adalimumab/MTX n=268 (95% confidence interval) | p-valuea | p-valueb | p-valuec | |
|---|---|---|---|---|---|---|
| Total Sharp Score | 5.7 (4.2-7.3) | 3.0 (1.7-4.3) | 1.3 (0.5-2.1) | <0.001 | 0.0020 | <0.001 |
| Erosion score | 3.7 (2.7-4.7) | 1.7 (1.0-2.4) | 0.8 (0.4-1.2) | <0.001 | 0.0082 | <0.001 |
| JSN score | 2.0 (1.2-2.8) | 1.3 (0.5-2.1) | 0.5 (0-1.0) | <0.001 | 0.0037 | 0.151 |
a p-value is from the pairwise comparison of methotrexate monotherapy and adalimumab/methotrexate combination therapy using the Mann-Whitney U test.
b p-value is from the pairwise comparison of adalimumab monotherapy and adalimumab/methotrexate combination therapy using the Mann-Whitney U test
c p-value is from the pairwise comparison of adalimumab monotherapy and methotrexate monotherapy using the Mann-Whitney U test
Following 52 weeks and 104 weeks of treatment, the percentage of patients without progression (change from baseline in modified Total Sharp Score ≤0.5) was significantly higher with adalimumab/methotrexate combination therapy (63.8% and 61.2% respectively) compared to methotrexate monotherapy (37.4% and 33.5% respectively, p<0.001) and adalimumab monotherapy (50.7%, p<0.002 and 44.5%, p<0.001 respectively).
In the open-label extension of RA study V, the mean change from baseline at Year 10 in the modified Total Sharp Score was 10.8, 9.2 and 3.9 in patients originally randomised to methotrexate monotherapy, adalimumab monotherapy and adalimumab/methotrexate combination therapy, respectively. The corresponding proportions of patients with no radiographic progression were 31.3%, 23.7% and 36.7% respectively.
Health-related quality of life and physical function were assessed using the disability index of the Health Assessment Questionnaire (HAQ) in the four original adequate and well-controlled trials, which was a pre-specified primary endpoint at Week 52 in RA study III. All doses/schedules of adalimumab in all four studies showed statistically significantly greater improvement in the disability index of the HAQ from baseline to Month 6 compared to placebo and in RA study III the same was seen at Week 52. Results from the Short Form Health Survey (SF 36) for all doses/schedules of adalimumab in all four studies support these findings, with statistically significant physical component summary (PCS) scores, as well as statistically significant pain and vitality domain scores for the 40 mg every other week dose. A statistically significant decrease in fatigue as measured by functional assessment of chronic illness therapy (FACIT) scores was seen in all three studies in which it was assessed (RA studies I, III, IV).
In RA study III, most subjects who achieved improvement in physical function and continued treatment maintained improvement through Week 520 (120 months) of open-label treatment. Improvement in quality of life was measured up to Week 156 (36 months) and improvement was maintained through that time.
In RA study V, the improvement in the HAQ disability index and the physical component of the SF 36 showed greater improvement (p<0.001) for adalimumab/methotrexate combination therapy versus methotrexate monotherapy and adalimumab monotherapy at Week 52, which was maintained through Week 104. Among the 250 subjects who completed the open-label extension study, improvements in physical function were maintained through 10 years of treatment.
For the pooled crossover RA studies VI and VII, a statistically significant difference for injection site pain immediately after dosing was observed between 40 mg/0.8 ml adalimumab and 40 mg/0.4 ml adalimumab (mean VAS of 3.7 cm versus 1.2 cm, scale of 0-10 cm, p<0.001). This represented an 84% median reduction in injection site pain.
The safety and efficacy of adalimumab were studied in adult patients with chronic plaque psoriasis (≥10% BSA involvement and Psoriasis Area and Severity Index (PASI) ≥12 or ≥10) who were candidates for systemic therapy or phototherapy in randomised, double-blind studies. 73% of patients enroled in Psoriasis Studies I and II had received prior systemic therapy or phototherapy. The safety and efficacy of adalimumab were also studied in adult patients with moderate to severe chronic plaque psoriasis with concomitant hand and/or foot psoriasis who were candidates for systemic therapy in a randomised double-blind study (Psoriasis Study III).
Psoriasis Study I (REVEAL) evaluated 1,212 patients within three treatment periods. In period A, patients received placebo or adalimumab at an initial dose of 80 mg followed by 40 mg every other week starting one week after the initial dose. After 16 weeks of therapy, patients who achieved at least a PASI 75 response (PASI score improvement of at least 75% relative to baseline), entered period B and received open-label 40 mg adalimumab every other week. Patients who maintained ≥ PASI 75 response at Week 33 and were originally randomised to active therapy in Period A, were rerandomised in period C to receive 40 mg adalimumab every other week or placebo for an additional 19 weeks. Across all treatment groups, the mean baseline PASI score was 18.9 and the baseline Physician’s Global Assessment (PGA) score ranged from “moderate” (53% of subjects included) to “severe” (41%) to “very severe” (6%).
Psoriasis Study II (CHAMPION) compared the efficacy and safety of adalimumab versus methotrexate and placebo in 271 patients. Patients received placebo, an initial dose of MTX 7.5 mg and thereafter dose increases up to Week 12, with a maximum dose of 25 mg or an initial dose of 80 mg adalimumab followed by 40 mg every other week (starting one week after the initial dose) for 16 weeks. There are no data available comparing adalimumab and MTX beyond 16 weeks of therapy. Patients receiving MTX who achieved a ≥ PASI 50 response at Week 8 and/or 12 did not receive further dose increases. Across all treatment groups, the mean baseline PASI score was 19.7 and the baseline PGA score ranged from “mild” (<1%) to “moderate” (48%) to “severe” (46%) to “very severe” (6%).
Patients participating in all Phase 2 and Phase 3 psoriasis studies were eligible to enrol into an openlabel extension trial, where adalimumab was given for at least an additional 108 weeks.
In Psoriasis Studies I and II, a primary endpoint was the proportion of patients who achieved a PASI 75 response from baseline at Week 16 (see Tables 9 and 10).
Table 9. Ps study I (REVEAL) - Efficacy results at 16 weeks:
| Placebo N=398 n (%) | Adalimumab 40 mg eow N=814 n (%) | |
|---|---|---|
| ≥ PASI 75a | 26 (6.5) | 578 (70.9)b |
| PASI 100 | 3 (0.8) | 163 (20.0)b |
| PGA: Clear/minimal | 17 (4.3) | 506 (62.2)b |
a Percent of patients achieving PASI75 response was calculated as centre-adjusted rate
b p<0.001, adalimumab vs. placebo
Table 10. Ps study II (CHAMPION) - Efficacy results at 16 weeks:
| Placebo N=53 n (%) | MTX N=110 n (%) | Adalimumab 40 mg eow N=108 n (%) | |
|---|---|---|---|
| ≥ PASI 75 | 10 (18.9) | 39 (35.5) | 86 (79.6)a,b |
| PASI 100 | 1 (1.9) | 8 (7.3) | 18 (16.7)c,d |
| PGA: Clear/minimal | 6 (11.3) | 33 (30.0) | 79 (73.1)a,b |
a p<0.001 adalimumab vs. placebo
b p<0.001 adalimumab vs. methotrexate
c p<0.01 adalimumab vs. placebo
d p<0.05 adalimumab vs. methotrexate
In Psoriasis Study I, 28% of patients who were PASI 75 responders and were re-randomised to placebo at Week 33 compared to 5% continuing on adalimumab, p<0.001, experienced “loss of adequate response” (PASI score after Week 33 and on or before Week 52 that resulted in a < PASI 50 response relative to baseline with a minimum of a 6-point increase in PASI score relative to Week 33). Of the patients who lost adequate response after re-randomisation to placebo who then enroled into the open-label extension trial, 38% (25/66) and 55% (36/66) regained PASI 75 response after 12 and 24 weeks of re-treatment, respectively.
A total of 233 PASI 75 responders at Week 16 and Week 33 received continuous adalimumab therapy for 52 weeks in Psoriasis Study I, and continued adalimumab in the open-label extension trial. PASI 75 and PGA of clear or minimal response rates in these patients were 74.7% and 59.0%, respectively, after an additional 108 weeks of open-label therapy (total of 160 weeks). In an analysis in which all patients who dropped out of the study for adverse events or lack of efficacy, or who dose-escalated, were considered non-responders, PASI 75 and PGA of clear or minimal response rates in these patients were 69.6% and 55.7%, respectively, after an additional 108 weeks of open-label therapy (total of 160 weeks).
A total of 347 stable responders participated in a withdrawal and retreatment evaluation in an openlabel extension study. During the withdrawal period, symptoms of psoriasis returned over time with a median time to relapse (decline to PGA “moderate” or worse) of approximately 5 months. None of these patients experienced rebound during the withdrawal period. A total of 76.5% (218/285) of patients who entered the retreatment period had a response of PGA “clear” or “minimal” after 16 weeks of retreatment, irrespective of whether they relapsed during withdrawal (69.1%[123/178] and 88.8% [95/107] for patients who relapsed and who did not relapse during the withdrawal period, respectively). A similar safety profile was observed during retreatment as before withdrawal.
Significant improvements at Week 16 from baseline compared to placebo (Studies I and II) and MTX (Study II) were demonstrated in the DLQI (Dermatology Life Quality Index). In Study I, improvements in the physical and mental component summary scores of the SF-36 were also significant compared to placebo.
In an open-label extension study, for patients who dose escalated from 40 mg every other week (eow) to 40 mg weekly due to a PASI response below 50%, 26.4% (92/349) and 37.8% (132/349) of patients achieved PASI 75 response at Week 12 and 24, respectively.
Psoriasis Study III (REACH) compared the efficacy and safety of adalimumab versus placebo in 72 patients with moderate to severe chronic plaque psoriasis and hand and/or foot psoriasis. Patients received an initial dose of 80 mg adalimumab followed by 40 mg every other week (starting one week after the initial dose) or placebo for 16 weeks. At Week 16, a statistically significantly greater proportion of patients who received adalimumab achieved PGA of ‘clear’ or ‘almost clear’ for the hands and/or feet compared to patients who received placebo (30.6% versus 4.3%, respectively [p=0.014]).
Psoriasis Study IV compared efficacy and safety of adalimumab versus placebo in 217 adult patients with moderate to severe nail psoriasis. Patients received an initial dose of 80 mg adalimumab followed by 40 mg every other week (starting one week after the initial dose) or placebo for 26 weeks followed by open-label adalimumab treatment for an additional 26 weeks. Nail psoriasis assessments included the Modified Nail Psoriasis Severity Index (mNAPSI), the Physician’s Global Assessment of Fingernail Psoriasis (PGA-F) and the Nail Psoriasis Severity Index (NAPSI) (see Table 11). Adalimumab demonstrated a treatment benefit in nail psoriasis patients with different extents of skin involvement (BSA ≥10% (60% of patients) and BSA <10% and ≥5% (40% of patients)).
Table 11. Ps study IV – Efficacy results at 16, 26 and 52 weeks:
| Endpoint | Week 16 Placebo-Controlled | Week 26 Placebo-Controlled | Week 52 Open-label | |||
|---|---|---|---|---|---|---|
| Placebo N=108 | Adalimumab 40 mg eow N=109 | Placebo N=108 | Adalimumab 40 mg eow N=109 | Adalimumab 40 mg eow N=80 | ||
| ≥ mNAPSI 75 (%) | 2.9 | 26.0a | 3.4 | 46.6a | 65.0 | |
| PGA-F clear/minimal and ≥ 2-grade improvement (%) | 2.9 | 29.7a | 6.9 | 48.9a | 61.3 | |
| Percent Change in Total Fingernail NAPSI (%) | -7.8 | -44.2a | -11.5 | -56.2a | -72.2 | |
a p<0.001, adalimumab vs. placebo
Adalimumab-treated patients showed statistically significant improvements at Week 26 compared with placebo in the DLQI.
The safety and efficacy of adalimumab were assessed in randomised, double-blind, placebo-controlled studies and an open-label extension study in adult patients with moderate to severe HS who were intolerant, had a contraindication or an inadequate response to at least a 3-month trial of systemic antibiotic therapy. The patients in HS-I and HS-II had Hurley Stage II or III disease with at least 3 abscesses or inflammatory nodules.
Study HS-I (PIONEER I) evaluated 307 patients with 2 treatment periods. In Period A, patients received placebo or adalimumab at an initial dose of 160 mg at Week 0, 80 mg at Week 2, and 40 mg every week starting at Week 4 to Week 11. Concomitant antibiotic use was not allowed during the study. After 12 weeks of therapy, patients who had received adalimumab in Period A were rerandomised in Period B to 1 of 3 treatment groups (adalimumab 40 mg every week, adalimumab 40 mg every other week, or placebo from Week 12 to Week 35). Patients who had been randomised to placebo in Period A were assigned to receive adalimumab 40 mg every week in Period B.
Study HS-II (PIONEER II) evaluated 326 patients with 2 treatment periods. In Period A, patients received placebo or adalimumab at an initial dose of 160 mg at Week 0 and 80 mg at Week 2 and 40 mg every week starting at Week 4 to Week 11. 19.3% of patients had continued baseline oral antibiotic therapy during the study. After 12 weeks of therapy, patients who had received adalimumab in Period A were re-randomised in Period B to 1 of 3 treatment groups (adalimumab 40 mg every week, adalimumab 40 mg every other week, or placebo from Week 12 to Week 35). Patients who had been randomised to placebo in Period A were assigned to receive placebo in Period B.
Patients participating in Studies HS-I and HS-II were eligible to enrol into an open-label extension study in which adalimumab 40mg was administered every week. Mean exposure in all adalimumab population was 762 days. Throughout all 3 studies patients used topical antiseptic wash daily.
Reduction of inflammatory lesions and prevention of worsening of abscesses and draining fistulas was assessed using Hidradenitis Suppurativa Clinical Response (HiSCR; at least a 50% reduction in total abscess and inflammatory nodule count with no increase in abscess count and no increase in draining fistula count relative to Baseline). Reduction in HS-related skin pain was assessed using a Numeric Rating Scale in patients who entered the study with an initial baseline score of 3 or greater on a 11 point scale.
At Week 12, a significantly higher proportion of patients treated with adalimumab versus placebo achieved HiSCR. At Week 12, a significantly higher proportion of patients in Study HS-II experienced a clinically relevant decrease in HS-related skin pain (see Table 12). Patients treated with adalimumab had significantly reduced risk of disease flare during the initial 12 weeks of treatment.
Table 12. Efficacy results at 12 weeks, HS studies I and II:
| HS Study I | HS Study II | |||
|---|---|---|---|---|
| Placebo | Adalimumab 40 mg weekly | Placebo | Adalimumab 40 mg weekly | |
| Hidradenitis Suppurativa Clinical Response (HiSCR)a | N=154 40 (26.0%) | N=153 64 (41.8%)* | N=163 45 (27.6%) | N=163 96 (58.9%)*** |
| ≥ 30% Reduction in Skin Painb | N=109 27 (24.8%) | N=122 34 (27.9%) | N=111 23 (20.7%) | N=105 48 (45.7%)*** |
* p<0.05, ***p<0.001, adalimumab versus placebo
a Among all randomised patients.
b Among patients with baseline HS-related skin pain assessment ≥3, based on Numeric Rating Scale 0 – 10; 0 = no skin pain, 10 = skin pain as bad as you can imagine.
Treatment with adalimumab 40 mg every week significantly reduced the risk of worsening of abscesses and draining fistulas. Approximately twice the proportion of patients in the placebo group in the first 12 weeks of Studies HS-I and HS-II, compared with those in the adalimumab group experienced worsening of abscesses (23.0% vs 11.4%, respectively) and draining fistulas (30.0% vs 13.9%, respectively).
Greater improvements at Week 12 from baseline compared to placebo were demonstrated in skinspecific health-related quality of life, as measured by the Dermatology Life Quality Index (DLQI; Studies HS-I and HS-II), patient global satisfaction with medicinal product treatment as measured by the Treatment Satisfaction Questionnaire – medicinal products (TSQM; Studies HS-I and HS-II), and physical health as measured by the physical component summary score of the SF-36 (Study HS-I).
In patients with at least a partial response to adalimumab 40 mg weekly at Week 12, the HiSCR rate at Week 36 was higher in patients who continued weekly adalimumab than in patients in whom dosing frequency was reduced to every other week, or in whom treatment was withdrawn (see Table 13).
Table 13: Proportion of patientsa achieving HiSCRb at Weeks 24 and 36 after treatment reassignment from weekly adalimumab at Week 12:
| Placebo (treatment withdrawal) N=73 | Adalimumab 40 mg every other week N=70 | Adalimumab 40 mg weekly N=70 | |
|---|---|---|---|
| Week 24 | 24 (32.9%) | 36 (51.4%) | 40 (57.1%) |
| Week 36 | 22 (30.1%) | 28 (40.0%) | 39 (55.7%) |
a Patients with at least a partial response to adalimumab 40 mg weekly after 12 weeks of treatment.
b Patients meeting protocol-specified criteria for loss of response or no improvement were required to discontinue from the studies and were counted as non-responders.
Among patients who were at least partial responders at Week 12, and who received continuous weekly adalimumab therapy, the HiSCR rate at Week 48 was 68.3% and at Week 96 was 65.1%. Longer term treatment with adalimumab 40 mg weekly for 96 weeks identified no new safety findings.
Among patients whose adalimumab treatment was withdrawn at Week 12 in Studies HS-I and HS-II, the HiSCR rate 12 weeks after re-introduction of adalimumab 40 mg weekly returned to levels similar to that observed before withdrawal (56.0%).
The safety and efficacy of adalimumab were assessed in over 1,500 patients with moderately to severely active Crohn’s disease (Crohn’s Disease Activity Index (CDAI) ≥220 and ≤450) in randomised, double-blind, placebo-controlled studies. Concomitant stable doses of aminosalicylates, corticosteroids, and/or immunomodulatory agents were permitted and 80% of patients continued to receive at least one of these medicinal poroducts.
Induction of clinical remission (defined as CDAI <150) was evaluated in two studies, CD Study I (CLASSIC I) and CD Study II (GAIN). In CD Study I, 299 TNF-antagonist naive patients were randomised to one of four treatment groups; placebo at Weeks 0 and 2, 160 mg adalimumab at Week 0 and 80 mg at Week 2, 80 mg at Week 0 and 40 mg at Week 2, and 40 mg at Week 0 and 20 mg at Week 2. In CD Study II, 325 patients who had lost response or were intolerant to infliximab were randomised to receive either 160 mg adalimumab at Week 0 and 80 mg at Week 2 or placebo at Weeks 0 and 2. The primary non-responders were excluded from the studies and therefore these patients were not further evaluated.
Maintenance of clinical remission was evaluated in CD study III (CHARM). In CD Study III, 854 patients received open-label 80 mg at Week 0 and 40 mg at Week 2. At Week 4 patients were randomised to 40 mg every other week, 40 mg every week, or placebo with a total study duration of 56 weeks. Patients in clinical response (decrease in CDAI ≥70) at Week 4 were stratified and analysed separately from those not in clinical response at Week 4. Corticosteroid taper was permitted after Week 8.
CD study I and CD study II induction of remission and response rates are presented in Table 14.
Table 14. Induction of clinical remission and response (percent of patients):
| CD Study I: Infliximab Naive Patients | CD Study II: Infliximab Experienced Patients | ||||
|---|---|---|---|---|---|
| Placebo N=74 | Adalimumab 80/40 mg N=75 | Adalimumab 160/80 mg N=76 | Placebo N=166 | Adalimumab 160/80 mg N=159 | |
| Week 4 | |||||
| Clinical remission | 12% | 24% | 36%* | 7% | 21%* |
| Clinical response (CR-100) | 24% | 37% | 49%** | 25% | 38%** |
All p-values are pairwise comparisons of proportions for adalimumab versus placebo
* p<0.001
** p<0.01
Similar remission rates were observed for the 160/80 mg and 80/40 mg induction regimens by Week 8 and adverse events were more frequently noted in the 160/80 mg group.
In CD Study III, at Week 4, 58% (499/854) of patients were in clinical response and were assessed in the primary analysis. Of those in clinical response at Week 4, 48% had been previously exposed to other TNF-antagonists. Maintenance of remission and response rates are presented in Table 15. Clinical remission results remained relatively constant irrespective of previous TNF-antagonist exposure.
Disease-related hospitalisations and surgeries were statistically significantly reduced with adalimumab compared with placebo at Week 56.
Table 15. Maintenance of clinical remission and response (percent of patients):
| Placebo | Adalimumab 40 mg every other week | Adalimumab 40 mg every week | |
|---|---|---|---|
| Week 26 | N=170 | N=172 | N=157 |
| Clinical remission | 17% | 40%* | 47%* |
| Clinical response (CR-100) | 27% | 52%* | 52%* |
| Patients in steroid-free remission for ≥90 daysa | 3% (2/66) | 19% (11/58)** | 15% (11/74)** |
| Week 56 | N=170 | N=172 | N=157 |
| Clinical remission | 12% | 36%* | 41%* |
| Clinical response (CR-100) | 17% | 41%* | 48%* |
| Patients in steroid-free remission for ≥90 daysa | 5% (3/66) | 29% (17/58)* | 20% (15/74)** |
* p<0.001 for adalimumab versus placebo pairwise comparisons of proportions
** p<0.02 for adalimumab versus placebo pairwise comparisons of proportions a Of those receiving corticosteroids at baseline
Among patients who were not in response at Week 4, 43% of adalimumab maintenance patients responded by Week 12 compared to 30% of placebo maintenance patients. These results suggest that some patients who have not responded by Week 4 benefit from continued maintenance therapy through Week 12. Therapy continued beyond 12 weeks did not result in significantly more responses (see section 4.2).
117/276 patients from CD study I and 272/777 patients from CD studies II and III were followed through at least 3 years of open-label adalimumab therapy. 88 and 189 patients, respectively, continued to be in clinical remission. Clinical response (CR-100) was maintained in 102 and 233 patients, respectively.
In CD Study I and CD Study II, statistically significant improvement in the disease-specific inflammatory bowel disease questionnaire (IBDQ) total score was achieved at Week 4 in patients randomised to adalimumab 80/40 mg and 160/80 mg compared to placebo and was seen at Weeks 26 and 56 in CD Study III as well among the adalimumab treatment groups compared to the placebo group.
The safety and efficacy of multiple doses of adalimumab were assessed in adult patients with moderately to severely active ulcerative colitis (Mayo score 6 to 12 with endoscopy subscore of 2 to 3) in randomised, double-blind, placebo-controlled studies.
In study UC-I, 390 TNF-antagonist naïve patients were randomised to receive either placebo at Weeks 0 and 2, 160 mg adalimumab at Week 0 followed by 80 mg at Week 2, or 80 mg adalimumab at Week 0 followed by 40 mg at Week 2. After Week 2, patients in both adalimumab arms received 40 mg eow. Clinical remission (defined as Mayo score ≤2 with no subscore >1) was assessed at Week 8.
In study UC-II, 248 patients received 160 mg of adalimumab at Week 0, 80 mg at Week 2 and 40 mg eow thereafter, and 246 patients received placebo. Clinical results were assessed for induction of remission at Week 8 and for maintenance of remission at Week 52.
Patients induced with 160/80 mg adalimumab achieved clinical remission versus placebo at Week 8 in statistically significantly greater percentages in study UC-I (18% vs. 9% respectively, p=0.031) and study UC-II (17% vs. 9% respectively, p=0.019). In study UC-II, among those treated with adalimumab who were in remission at Week 8, 21/41 (51%) were in remission at Week 52.
Results from the overall UC-II study population are shown in Table 16.
Table 16. Response, remission and mucosal healing in study UC-II (percent of patients):
| Placebo | Adalimumab 40 mg eow | |
|---|---|---|
| Week 52 | N=246 | N=248 |
| Clinical response | 18% | 30%* |
| Clinical remission | 9% | 17%* |
| Mucosal healing | 15% | 25%* |
| Steroid-free remission for ≥90 daysa | 6% | 13%* |
| (N=140) | (N=150) | |
| Week 8 and 52 | ||
| Sustained response | 12% | 24%** |
| Sustained remission | 4% | 8%* |
| Sustained mucosal healing | 11% | 19%* |
Clinical remission is Mayo score ≤2 with no subscore >1;
Clinical response is decrease from baseline in Mayo score ≥3 points and ≥30% plus a decrease in the rectal bleeding subscore [RBS] ≥1 or an absolute RBS of 0 or 1;
* p<0.05 for adalimumab vs. placebo pairwise comparison of proportions
** p<0.001 for adalimumab vs. placebo pairwise comparison of proportions
a Of those receiving corticosteroids at baseline
Of those patients who had a response at Week 8, 47% were in response, 29% were in remission, 41% had mucosal healing, and 20% were in steroid-free remission for ≥90 days at Week 52.
Approximately 40% of patients in study UC-II had failed prior anti-TNF treatment with infliximab. The efficacy of adalimumab in those patients was reduced compared to that in anti-TNF naïve patients. Among patients who had failed prior anti-TNF treatment, Week 52 remission was achieved by 3% on placebo and 10% on adalimumab.
Patients from studies UC-I and UC-II had the option to roll over into an open-label long-term extension study (UC III). Following 3 years of adalimumab therapy, 75% (301/402) continued to be in clinical remission per partial Mayo score.
During 52 weeks of studies UC-I and UC-II, lower rates of all-cause hospitalisations and UC-related hospitalisations were observed for the adalimumab-treated arm compared to the placebo arm. The number of all cause hospitalisations in the adalimumab treatment group was 0.18 per patient year vs. 0.26 per patient year in the placebo group and the corresponding figures for UC-related hospitalisations were 0.12 per patient year vs. 0.22 per patient year.
In study UC-II, treatment with adalimumab resulted in improvements in the Inflammatory Bowel Disease Questionnaire (IBDQ) score.
The safety and efficacy of adalimumab were assessed in adult patients with non-infectious intermediate, posterior, and panuveitis, excluding patients with isolated anterior uveitis, in two randomised, double-masked, placebo-controlled studies (UV I and II). Patients received placebo or adalimumab at an initial dose of 80 mg followed by 40 mg every other week starting one week after the initial dose. Concomitant stable doses of one non-biologic immunosuppressant were permitted.
Study UV I evaluated 217 patients with active uveitis despite treatment with corticosteroids (oral prednisone at a dose of 10 to 60 mg/day). All patients received a 2-week standardised dose of prednisone 60 mg/day at study entry followed by a mandatory taper schedule, with complete corticosteroid discontinuation by Week 15.
Study UV II evaluated 226 patients with inactive uveitis requiring chronic corticosteroid treatment (oral prednisone 10 to 35 mg/day) at baseline to control their disease. Patients subsequently underwent a mandatory taper schedule, with complete corticosteroid discontinuation by Week 19.
The primary efficacy endpoint in both studies was ´time to treatment failure´. Treatment failure was defined by a multi-component outcome based on inflammatory chorioretinal and/or inflammatory retinal vascular lesions, anterior chamber (AC) cell grade, vitreous haze (VH) grade and best corrected visual acuity (BCVA).
Patients who completed Studies UV I and UV II were eligible to enrol in an uncontrolled long-term extension study with an originally planned duration of 78 weeks. Patients were allowed to continue on study medication beyond Week 78 until they had access to adalimumab.
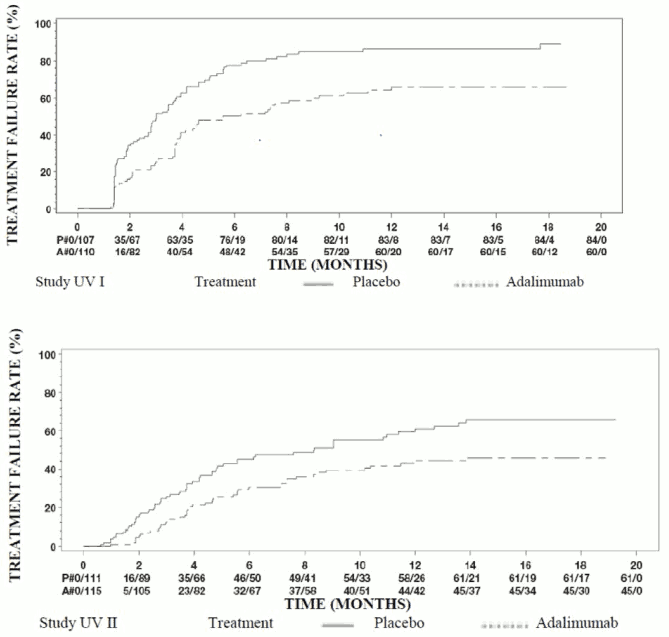
Results from both studies demonstrated statistically significant reduction of the risk of treatment failure in patients treated with adalimumab versus patients receiving placebo (see Table 17). Both studies demonstrated an early and sustained effect of adalimumab on the treatment failure rate versus placebo (see Figure 1).
Table 17. Time to treatment failure in studies UV I and UV II:
| Analysis Treatment | N | Failure N (%) | Median Time to Failure (months) | HRa | CI 95% for HRa | p valueb |
|---|---|---|---|---|---|---|
| Time to treatment failure at or after Week 6 in study UV I | ||||||
| Primary analysis (ITT) | ||||||
| Placebo | 107 | 84 (78.5) | 3.0 | -- | -- | -- |
| Adalimumab | 110 | 60 (54.5) | 5.6 | 0.50 | 0.36, 0.70 | <0.001 |
| TTime to treatment failure at or after Week 2 in study UV II | ||||||
| Primary analysis (ITT) | ||||||
| Placebo | 111 | 61 (55.0) | 8.3 | -- | -- | -- |
| Adalimumab | 115 | 45 (39.1) | NEc | 0.57 | 0.39, 0.84 | 0.004 |
Note: Treatment failure at or after Week 6 (Study UV I), or at or after Week 2 (Study UV II), was counted as event. Drop outs due to reasons other than treatment failure were censored at the time of dropping out.
a HR of adalimumab vs placebo from proportional hazards regression with treatment as factor.
b 2-sided p value from log rank test.
c NE = not estimable. Fewer than half of at-risk subjects had an event.
Figure 1. Kaplan-Meier curves summarising time to treatment failure on or after Week 6 (study UV I) or Week 2 (study UV II):
Note: P# = Placebo (Number of Events/Number at Risk); A# = Adalimumab (Number of Events/Number at Risk).
In Study UV I statistically significant differences in favour of adalimumab versus placebo were observed for each component of treatment failure. In Study UV II, statistically significant differences were observed for visual acuity only, but the other components were numerically in favour of adalimumab.
Of the 424 subjects included in the uncontrolled long-term extension of Studies UV I and UV II, 60 subjects were regarded ineligible (e.g. due to deviations or due to complications secondary to diabetic retinopathy, due to cataract surgery or vitrectomy) and were excluded from the primary analysis of efficacy. Of the 364 remaining patients, 269 evaluable patients (74%) reached 78 weeks of open-label adalimumab treatment. Based on the observed data approach, 216 (80.3%) were in quiescence (no active inflammatory lesions, AC cell grade ≤0.5+, VH grade ≤0.5+) with a concomitant steroid dose ≤7.5 mg per day, and 178 (66.2%) were in steroid-free quiescence. BCVA was either improved or maintained (<5 letters deterioration) in 88.6% of the eyes at week 78. Data beyond Week 78 were generally consistent with these results but the number of enroled subjects declined after this time. Overall, among the patients who discontinued the study, 18% discontinued due to adverse events, and 8% due to insufficient response to adalimumab treatment.
Patient reported outcomes regarding vision-related functioning were measured in both clinical studies, using the NEI VFQ-25. Adalimumab was numerically favoured for the majority of subscores with statistically significant mean differences for general vision, ocular pain, near vision, mental health, and total score in Study UV I, and for general vision and mental health in Study UV II. Vision related effects were not numerically in favour of adalimumab for colour vision in Study UVI and for colour vision, peripheral vision and near vision in Study UV II.
Anti-adalimumab antibodies may develop during adalimumab treatment. Formation of antiadalimumab antibodies is associated with increased clearance and reduced efficacy of adalimumab. There is no apparent correlation between the presence of anti-adalimumab antibodies and the occurrence of adverse events.
There are no clinical trials with adalimumab in adolescent patients with HS. Efficacy of adalimumab for the treatment of adolescent patients with HS is predicted based on the demonstrated efficacy and exposure-response relationship in adult HS patients and the likelihood that the disease course, pathophysiology, and active substance effects are substantially similar to that of adults at the same exposure levels. Safety of the recommended adalimumab dose in the adolescent HS population is based on cross-indication safety profile of adalimumab in both adults and paediatric patients at similar or more frequent doses (see section 5.2).
Adalimumab was assessed in a multicentre, randomised, double-blind clinical trial designed to evaluate the efficacy and safety of induction and maintenance treatment with doses dependent on body weight (<40 kg or ≥40 kg) in 192 paediatric subjects between the ages of 6 and 17 (inclusive) years, with moderate to severe Crohn’s disease (CD) defined as Paediatric Crohn’s Disease Activity Index (PCDAI) score >30. Subjects had to have failed conventional therapy (including a corticosteroid and/or an immunomodulator) for CD. Subjects may also have previously lost response or been intolerant to infliximab.
All subjects received open-label induction therapy at a dose based on their Baseline body weight: 160 mg at Week 0 and 80 mg at Week 2 for subjects ≥40 kg, and 80 mg and 40 mg, respectively, for subjects <40 kg.
At Week 4, subjects were randomised 1:1 based on their body weight at the time to either the Low Dose or Standard Dose maintenance regimens as shown in Table 18.
Table 18. Maintenance regimen:
| Patient Weight | Low dose | Standard dose |
|---|---|---|
| <40 kg | 10 mg eow | 20 mg eow |
| ≥40 kg | 20 mg eow | 40 mg eow |
The primary endpoint of the study was clinical remission at Week 26, defined as PCDAI score ≤10.
Clinical remission and clinical response (defined as reduction in PCDAI score of at least 15 points from Baseline) rates are presented in Table 19. Rates of discontinuation of corticosteroids or immunomodulators are presented in Table 20.
Table 19. Paediatric CD study, PCDAI clinical remission and response:
| Standard dose 40/20 mg eow N=93 | Low dose 20/10 mg eow N=95 | p value* | |
|---|---|---|---|
| Week 26 | |||
| Clinical remission | 38.7% | 28.4% | 0.075 |
| Clinical response | 59.1% | 48.4% | 0.073 |
| Week 52 | |||
| Clinical remission | 33.3% | 23.2% | 0.100 |
| Clinical response | 41.9% | 28.4% | 0.038 |
* p value for Standard Dose versus Low Dose comparison
Table 20. Paediatric CD study, discontinuation of corticosteroids or immunomodulators and fistula remission:
| Standard dose 40/20 mg eow | Low dose 20/10 mg eow | p value1 | |
|---|---|---|---|
| Discontinued corticosteroids | N=33 | N=38 | |
| Week 26 | 84.8% | 65.8% | 0.066 |
| Week 52 | 69.7% | 60.5% | 0.420 |
| Discontinuation of immunomodulators2 | N=60 | N=57 | |
| Week 52 | 30.0% | 29.8% | 0.983 |
| Fistula remission3 | N=15 | N=21 | |
| Week 26 | 46.7% | 38.1% | 0.608 |
| Week 52 | 40.0% | 23.8% | 0.303 |
1 p value for Standard Dose versus Low Dose comparison
2 Immunosuppressant therapy could only be discontinued at or after Week 26 at the investigator’s discretion if the subject met the clinical response criterion
3 defined as a closure of all fistulas that were draining at Baseline for at least 2 consecutive post-Baseline visits
Statistically significant increases (improvement) from Baseline to Week 26 and 52 in Body Mass Index and height velocity were observed for both treatment groups.
Statistically and clinically significant improvements from Baseline were also observed in both treatment groups for quality of life parameters (including IMPACT III).
One hundred patients (n=100) from the Paediatric CD Study continued in an open-label long-term extension study. After 5 years of adalimumab therapy, 74.0% (37/50) of the 50 patients remaining in the study continued to be in clinical remission, and 92.0% (46/50) of patients continued to be in clinical response per PCDAI.
The safety and efficacy of adalimumab was assessed in a multicentre, randomised, double-blind, trial in 93 paediatric patients from 5 to 17 years of age with moderate to severe ulcerative colitis (Mayo score 6 to 12 with endoscopy subscore of 2 to 3 points, confirmed by centrally read endoscopy) who had an inadequate response or intolerance to conventional therapy. Approximately 16% of patients in the study had failed prior anti-TNF treatment. Patients who received corticosteroids at enrolment were allowed to taper their corticosteroid therapy after Week 4.
In the induction period of the study, 77 patients were randomised 3:2 to receive double-blind treatment with adalimumab at an induction dose of 2.4 mg/kg (maximum of 160 mg) at Week 0 and Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2; or an induction dose of 2.4 mg/kg (maximum of 160 mg) at Week 0, placebo at Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2. Both groups received 0.6 mg/kg (maximum of 40 mg) at Week 4 and Week 6. Following an amendment to the study design, the remaining 16 patients who enroled in the induction period received open-label treatment with adalimumab at the induction dose of 2.4 mg/kg (maximum of 160 mg) at Week 0 and Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2.
At Week 8, 62 patients who demonstrated clinical response per Partial Mayo Score (PMS; defined as a decrease in PMS ≥ 2 points and ≥30% from Baseline) were randomised equally to receive doubleblind maintenance treatment with adalimumab at a dose of 0.6 mg/kg (maximum of 40 mg) every week (ew), or a maintenance dose of 0.6 mg/kg (maximum of 40 mg) every other week (eow). Prior to an amendment to the study design, 12 additional patients who demonstrated clinical response per PMS were randomised to receive placebo but were not included in the confirmatory analysis of efficacy.
Disease flare was defined as an increase in PMS of at least 3 points (for patients with PMS of 0 to 2 at Week 8), at least 2 points (for patients with PMS of 3 to 4 at Week 8), or at least 1 point (for patients with PMS of 5 to 6 at Week 8).
Patients who met criteria for disease flare at or after Week 12 were randomised to receive a reinduction dose of 2.4 mg/kg (maximum of 160 mg) or a dose of 0.6 mg/kg (maximum of 40 mg) and continued to receive their respective maintenance dose regimen afterwards.
The co-primary endpoints of the study were clinical remission per PMS (defined as PMS ≤2 and no individual subscore >1) at Week 8, and clinical remission per FMS (Full Mayo Score) (defined as a Mayo Score ≤2 and no individual subscore >1) at Week 52 in patients who achieved clinical response per PMS at Week 8.
Clinical remission rates per PMS at Week 8 for patients in each of the adalimumab double-blind induction groups are presented in Table 21.
Table 21. Clinical remission per PMS at 8 weeks:
| Adalimumaba Maximum of 160 mg at Week 0 / Placebo at Week 1 N=30 | Adalimumabb,c Maximum of 160 mg at Week 0 and Week 1 N=47 | |
|---|---|---|
| Clinical remission | 13/30 (43.3%) | 28/47 (59.6%) |
a Adalimumab 2.4 mg/kg (maximum of 160 mg) at Week 0, placebo at Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2
b Adalimumab 2.4 mg/kg (maximum of 160 mg) at Week 0 and Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2
c Not including open-label Induction dose of Adalimumab 2.4 mg/kg (maximum of 160 mg) at Week 0 and Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2
Note 1: Both induction groups received 0.6 mg/kg (maximum of 40 mg) at Week 4 and Week 6
Note 2: Patients with missing values at Week 8 were considered as not having met the endpoint
At Week 52, clinical remission per FMS in Week 8 responders, clinical response per FMS (defined as a decrease in Mayo Score ≥3 points and ≥30% from Baseline) in Week 8 responders, mucosal healing (defined as Mayo endoscopy subscore ≤1) in Week 8 responders, clinical remission per FMS in Week 8 remitters, and the proportion of subjects in corticosteroid-free remission per FMS in Week 8 responders were assessed in patients who received adalimumab at the double-blind maximum 40 mg eow (0.6 mg/kg) and maximum 40 mg ew (0.6 mg/kg) maintenance doses (Table 22).
Table 22. Efficacy results at 52 weeks:
| Adalimumaba Maximum of 40 mg eow N=31 | Adalimumabb Maximum of 40 mg ew N=31 | |
|---|---|---|
| Clinical remission in Week 8 PMS responders | 9/31 (29.0%) | 14/31 (45.2%) |
| Clinical response in Week 8 PMS responders | 19/31 (61.3%) | 21/31 (67.7%) |
| Mucosal healing in Week 8 PMS responders | 12/31 (38.7%) | 16/31 (51.6%) |
| Clinical remission in Week 8 PMS remitters | 9/21 (42.9%) | 10/22 (45.5%) |
| Corticosteroid-free remission in Week 8 PMS respondersc | 4/13 (30.8%) | 5/16 (31.3%) |
a Adalimumab 0.6 mg/kg (maximum of 40 mg) every other week
b Adalimumab 0.6 mg/kg (maximum of 40 mg) every week
c In patients receiving concomitant corticosteroids at baseline
Note: Patients with missing values at Week 52 or who were randomised to receive re-induction or maintenance treatment were considered non-responders for Week 52 endpoints
Additional exploratory efficacy endpoints included clinical response per the Paediatric Ulcerative Colitis Activity Index (PUCAI) (defined as a decrease in PUCAI ≥20 points from Baseline) and clinical remission per PUCAI (defined as PUCAI <10) at Week 8 and Week 52 (Table 23).
Table 23. Exploratory endpoints results per PUCAI:
| Week 8 | ||
| Adalimumaba Maximum of 160 mg at Week 0 / Placebo at Week 1 N=30 | Adalimumabb,c Maximum of 160 mg at Week 0 and Week 1 N=47 | |
| Clinical remission per PUCAI | 10/30 (33.3%) | 22/47 (46.8%) |
| Clinical response per PUCAI | 15/30 (50.0%) | 32/47 (68.1%) |
| Week 52 | ||
| Adalimumabd Maximum of 40 mg eow N=31 | Adalimumabe Maximum of 40 mg ew N=31 | |
| Clinical remission per PUCAI in Week 8 PMS responders | 14/31 (45.2%) | 18/31 (58.1%) |
| Clinical response per PUCAI in Week 8 PMS responders | 18/31 (58.1%) | 16/31 (51.6%) |
a Adalimumab 2.4 mg/kg (maximum of 160 mg) at Week 0, placebo at Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2
b Adalimumab 2.4 mg/kg (maximum of 160 mg) at Week 0 and Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2
c Not including open-label Induction dose of Adalimumab 2.4 mg/kg (maximum of 160 mg) at Week 0 and Week 1, and 1.2 mg/kg (maximum of 80 mg) at Week 2
d Adalimumab 0.6 mg/kg (maximum of 40 mg) every other week e Adalimumab 0.6 mg/kg (maximum of 40 mg) every week
Note 1: Both induction groups received 0.6 mg/kg (maximum of 40 mg) at Week 4 and Week 6
Note 2: Patients with missing values at Week 8 were considered as not having met the endpoints
Note 3: Patients with missing values at Week 52 or who were randomised to receive re-induction or maintenance treatment were considered non-responders for Week 52 endpoints
Of the adalimumab-treated patients who received re-induction treatment during the maintenance period, 2/6 (33%) achieved clinical response per FMS at Week 52.
Clinically meaningful improvements from baseline were observed in IMPACT III and the caregiver Work Productivity and Activity Impairment (WPAI) scores for the groups treated with adalimumab.
Clinically meaningful increases (improvement) from baseline in height velocity were observed for the groups treated with adalimumab, and clinically meaningful increases (improvement) from baseline in Body Mass Index were observed for subjects on the high maintenance dose of maximum 40 mg (0.6 mg/kg) ew.
The safety and efficacy of adalimumab was assessed in a randomised, double-masked, controlled study of 90 paediatric patients from 2 to <18 years of age with active JIA-associated non-infectious anterior uveitis who were refractory to at least 12 weeks of methotrexate treatment. Patients received either placebo or 20 mg adalimumab (if <30 kg) or 40 mg adalimumab (if ≥30 kg) every other week in combination with their baseline dose of methotrexate.
The primary endpoint was ‘time to treatment failure’. The criteria determining treatment failure were worsening or sustained non-improvement in ocular inflammation, partial improvement with development of sustained ocular co-morbidities or worsening of ocular co-morbidities, non-permitted use of concomitant medicinal products, and suspension of treatment for an extended period of time.
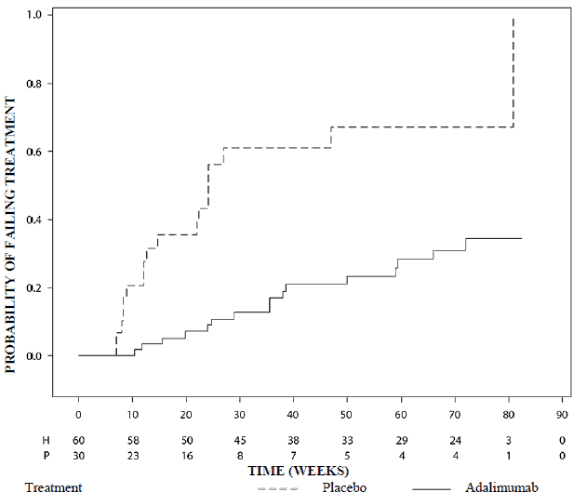
Adalimumab significantly delayed the time to treatment failure, as compared to placebo (see Figure 2, p<0.0001 from log rank test). The median time to treatment failure was 24.1 weeks for subjects treated with placebo, whereas the median time to treatment failure was not estimable for subjects treated with adalimumab because less than one-half of these subjects experienced treatment failure. Adalimumab significantly decreased the risk of treatment failure by 75% relative to placebo, as shown by the hazard ratio (HR = 0.25 [95% CI: 0.12, 0.49]).
Figure 2. Kaplan-Meier curves summarising time to treatment failure in the paediatric uveitis study:
Note: P = Placebo (Number at Risk); H = Adalimumab (Number at Risk).
After subcutaneous administration of a single 40 mg dose, absorption and distribution of adalimumab was slow, with peak serum concentrations being reached about 5 days after administration. The average absolute bioavailability of adalimumab estimated from three studies conducted with the reference product following a single 40 mg subcutaneous dose was 64%. After single intravenous doses ranging from 0.25 to 10 mg/kg, concentrations were dose proportional. After doses of 0.5 mg/kg (~40 mg), clearances ranged from 11 to 15 ml/hour, the distribution volume (Vss) ranged from 5 to 6 litres and the mean terminal phase half-life was approximately two weeks. Adalimumab concentrations in the synovial fluid from several rheumatoid arthritis patients ranged from 31-96% of those in serum.
Following subcutaneous administration of 40 mg of adalimumab every other week in adult rheumatoid arthritis (RA) patients the mean steady-state trough concentrations were approximately 5 μg/ml (without concomitant methotrexate) and 8 to 9 μg/ml (with concomitant methotrexate), respectively. The serum adalimumab trough levels at steady-state increased roughly proportionally with dose following 20, 40 and 80 mg subcutaneous dosing every other week and every week.
In adult patients with psoriasis, the mean steady-state trough concentration was 5 μg/ml during adalimumab 40 mg every other week monotherapy treatment.
In adult patients with HS, a dose of adalimumab 160 mg on Week 0 followed by 80 mg on Week 2 achieved serum adalimumab trough concentrations of approximately 7 to 8 μg/ml at Week 2 and Week 4. The mean steady-state trough concentration at Week 12 through Week 36 were approximately 8 to 10 μg/ml during adalimumab 40 mg every week treatment.
Adalimumab exposure in adolescent HS patients was predicted using population pharmacokinetic modelling and simulation based on cross-indication pharmacokinetics in other paediatric patients (paediatric psoriasis, juvenile idiopathic arthritis, paediatric Crohn’s disease, and enthesitis-related arthritis). The recommended adolescent HS dosing schedule is 40 mg every other week. Since exposure to adalimumab can be affected by body size, adolescents with higher body weight and inadequate response may benefit from receiving the recommended adult dose of 40 mg every week.
In patients with Crohn’s disease, the loading dose of 80 mg adalimumab on Week 0 followed by 40 mg adalimumab on Week 2 achieves serum adalimumab trough concentrations of approximately 5.5 μg/ml during the induction period. A loading dose of 160 mg adalimumab on Week 0 followed by 80 mg adalimumab on Week 2 achieves serum adalimumab trough concentrations of approximately 12 μg/ml during the induction period. Mean steady-state trough levels of approximately 7 μg/ml were observed in Crohn’s disease patients who received a maintenance dose of 40 mg adalimumab every other week.
In paediatric patients with moderate to severe CD, the open-label adalimumab induction dose was 160/80 mg or 80/40 mg at Weeks 0 and 2, respectively, dependent on a body weight cut-off of 40 kg. At Week 4, patients were randomised 1:1 to either the standard dose (40/20 mg every other week) or low dose (20/10 mg every other week) maintenance treatment groups based on their body weight. The mean (±SD) serum adalimumab trough concentrations achieved at Week 4 were 15.7 ± 6.6 μg/ml for patients ≥40 kg (160/80 mg) and 10.6 ± 6.1 μg/ml for patients <40 kg (80/40 mg).
For patients who stayed on their randomised therapy, the mean (±SD) adalimumab trough concentrations at Week 52 were 9.5 ± 5.6 μg/ml for the standard dose group and 3.5 ± 2.2 μg/ml for the low dose group. The mean trough concentrations were maintained in patients who continued to receive adalimumab treatment every other week for 52 weeks. For patients who dose escalated from every other week to weekly regimen, the mean (±SD) serum concentrations of adalimumab at Week 52 were 15.3 ± 11.4 μg/ml (40/20 mg, weekly) and 6.7 ± 3.5 μg/ml (20/10 mg, weekly).
In patients with ulcerative colitis, a loading dose of adalimumab 160 mg on Week 0 followed by adalimumab 80 mg on Week 2 achieves serum adalimumab trough concentrations of approximately 12 μg/ml during the induction period. Mean steady-state trough levels of approximately 8 μg/ml were observed in ulcerative colitis patients who received a maintenance dose of adalimumab 40 mg every other week.
Following the subcutaneous administration of body weight-based dosing of 0.6 mg/kg (maximum of 40 mg) every other week to paediatric patients with ulcerative colitis, the mean trough steady-state serum adalimumab concentration was 5.01±3.28 μg/ml at Week 52. For patients who received 0.6 mg/kg (maximum of 40 mg) every week, the mean (±SD) trough steady-state serum adalimumab concentration was 15.7±5.60 μg/ml at Week 52.
In adult patients with uveitis, a loading dose of adalimumab 80 mg on Week 0 followed by adalimumab 40 mg every other week starting at Week 1, resulted in mean steady-state concentrations of approximately 8 to 10 μg/ml.
Adalimumab exposure in paediatric uveitis patients was predicted using population pharmacokinetic modelling and simulation based on cross-indication pharmacokinetics in other paediatric patients (paediatric psoriasis, juvenile idiopathic arthritis, paediatric Crohn’s disease, and enthesitis-related arthritis). No clinical exposure data are available on the use of a loading dose in children <6 years. The predicted exposures indicate that in the absence of methotrexate, a loading dose may lead to an initial increase in systemic exposure.
Population pharmacokinetic and pharmacokinetic/pharmacodynamic modelling and simulation predicted comparable adalimumab exposure and efficacy in patients treated with 80 mg every other week when compared with 40 mg every week (including adult patients with RA, HS, UC, CD or Ps, patients with adolescent HS, and paediatric patients ≥40 kg with CD and UC).
On the basis of clinical trial data in patients with JIA (pJIA and ERA), an exposure-response relationship was established between plasma concentrations and PedACR 50 response. The apparent adalimumab plasma concentration that produces half the maximum probability of PedACR 50 response (EC50) was 3 μg/ml (95% CI: 1-6 μg/ml).
Exposure-response relationships between adalimumab concentration and efficacy in paediatric patients with severe chronic plaque psoriasis were established for PASI 75 and PGA clear or minimal, respectively. PASI 75 and PGA clear or minimal increased with increasing adalimumab concentrations, both with a similar apparent EC50 of approximately 4.5 μg/ml (95% CI 0.4-47.6 and 1.9-10.5, respectively).
Population pharmacokinetic analyses with data from over 1,300 RA patients revealed a trend toward higher apparent clearance of adalimumab with increasing body weight. After adjustment for weight differences, gender and age appeared to have a minimal effect on adalimumab clearance. The serum levels of free adalimumab (not bound to anti-adalimumab antibodies, AAA) were observed to be lower in patients with measurable AAA.
Adalimumab has not been studied in patients with hepatic or renal impairment.
Non-clinical data reveal no special hazard for humans based on studies of single dose toxicity, repeated dose toxicity, and genotoxicity.
An embryo-foetal developmental toxicity/perinatal developmental study has been performed in cynomolgus monkeys at 0, 30 and 100 mg/kg (9-17 monkeys/group) and has revealed no evidence of harm to the foetuses due to adalimumab. Neither carcinogenicity studies, nor a standard assessment of fertility and postnatal toxicity, were performed with adalimumab due to the lack of appropriate models for an antibody with limited cross-reactivity to rodent TNF and to the development of neutralising antibodies in rodents.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.