LOKELMA Powder for oral suspension Ref.[115591] Active ingredients: Sodium zirconium cyclosilicate
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: All other therapeutic products; Drugs for treatment of hyperkalaemia and hyperphosphatemia
ATC code: V03AE10
Mechanism of action
Sodium zirconium cyclosilicate is a non-absorbed, non-polymer inorganic powder with a uniform micropore structure that preferentially captures potassium in exchange for hydrogen and sodium cations. Sodium zirconium cyclosilicate is highly selective for potassium ions, even in the presence of other cations, such as calcium and magnesium, in vitro. Sodium zirconium cyclosilicate captures potassium throughout the entire gastrointestinal (GI) tract and reduces the concentration of free potassium in the GI lumen, thereby lowering serum potassium levels and increasing faecal potassium excretion to resolve hyperkalaemia.
Pharmacodynamic effects
Sodium zirconium cyclosilicate starts reducing serum potassium concentrations as soon as 1 hour after ingestion and normokalaemia can be achieved typically within 24 to 48 hours. Sodium zirconium cyclosilicate does not affect serum calcium or magnesium concentrations, or urinary sodium excretion. There is a close correlation between starting serum potassium levels and effect size; patients with higher starting serum potassium levels have greater reductions in serum potassium. There is a reduction in urinary potassium excretion which is a consequence of a reduction in serum potassium concentration. In a study of healthy subjects given Lokelma 5 g or 10 g once daily for four days, dose- dependent reduction in serum potassium concentration and total urinary potassium excretion were accompanied by mean increases in faecal potassium excretion. No statistically significant changes in urinary sodium excretion were observed.
There were no studies conducted to investigate the pharmacodynamics when sodium zirconium cyclosilicate is administered with or without food.
Sodium zirconium cyclosilicate has also been shown to bind ammonium in vitro and in vivo, thereby removing ammonium and increasing serum bicarbonate levels. Lokelma-treated patients experienced an increase of 1.1 mmol/L at 5 g once daily, 2.3 mmol/L at 10 g once daily and 2.6 mmol/L at 15 g once daily in bicarbonate compared with a mean increase of 0.6 mmol/L for those receiving placebo. In an environment where other factors affecting renin and aldosterone were not controlled, Lokelma demonstrated a dose-independent change in mean serum aldosterone levels (range: -30% to -31%) compared with the placebo group (+14%). No consistent effect on systolic and diastolic blood pressure has been observed.
In addition, mean reductions in blood urea nitrogen (BUN) were observed in the 5 g (1.1 mg/dL) and 10 g (2.0 mg/dL) three times daily groups compared with small mean increases in the placebo (0.8 mg/dL) and low dose sodium zirconium cyclosilicate (0.3 mg/dL) groups.
Clinical efficacy and safety
The potassium-lowering effects of Lokelma have been demonstrated in three randomised, double-blind, placebo-controlled trials in patients with hyperkalaemia. All three studies tested the initial effect of Lokelma to correct hyperkalaemia during a 48-hour period and two studies also tested maintenance of normokalaemia effect obtained. The maintenance studies included patients with chronic kidney disease (58%), heart failure (10%), diabetes mellitus (62%) and RAAS inhibitor therapy (68%). In addition, two open-label maintenance studies tested long-term safety of Lokelma. These five studies included 1 760 patients given doses of Lokelma; 507 exposed for at least 360 days. In addition, the efficacy and safety of Lokelma was studied in a double-blind, placebo-controlled trial of 196 chronic haemodialysis patients with hyperkalaemia, who received doses of Lokelma for 8 weeks. In the studies, Lokelma reduced serum potassium and maintained normal serum potassium levels regardless of the underlying cause of hyperkalaemia, age, sex, race, comorbid disease or concomitant use of RAAS inhibitors. No dietary restrictions were imposed; patients were instructed to continue their usual diet without any specified alterations.
Study 1
A two-phase, placebo-controlled correction and maintenance use study
A two-part, double-blind, randomised, placebo-controlled clinical trial of 753 patients (mean age of 66 years, range 22 to 93 years) with hyperkalaemia (5 to ≤6.5 mmol/L, baseline potassium average 5.3 mmol/L), and included patients with chronic kidney disease, heart failure, diabetes mellitus and those on RAAS inhibitor therapy.
During the correction phase, patients were randomised to receive Lokelma (1.25 g, 2.5 g, 5 g or 10 g) or placebo, administered three times daily for the initial 48 hours (Table 2).
Table 2. Correction phase (Study 1): Percentage of normokalaemic subjects after 48 hours of Lokelma:
| Lokelma dose (three times daily) | |||||
|---|---|---|---|---|---|
| Placebo | 1.25 g | 2.5 g | 5 g | 10 g | |
| N | 158 | 154 | 141 | 157 | 143 |
| Baseline serum potassium, mmol/L | 5.3 | 5.4 | 5.4 | 5.3 | 5.3 |
| Normokalaemic at 48 hours, % | 48 | 51 | 68 | 78 | 86 |
| p-value vs. placebo | NS | <0.001 | <0.001 | <0.001 | |
NS: not significant
Lokelma 10 g administered three times daily lowered serum potassium by 0.7 mmol/L at 48 hours (p<0.001 vs. placebo); statistically significant 14% potassium reduction was observed 1 hour after the first dose. Patients with higher starting potassium levels had a greater response to Lokelma. Patients with pre-treatment potassium levels in excess of 5.5 mmol/L (average baseline 5.8 mmol/L) saw an average decrease of 1.1 mmol/L at 48 hours while those with starting potassium levels at or below 5.3 mmol/L had an average decrease of 0.6 mmol/L at the highest dose.
Patients who became normokalaemic after receiving Lokelma during the correction phase were re-randomised to receive once daily placebo or once daily Lokelma at the same dose level as they had received three times daily during the correction phase (Table 3).
Table 3. Maintenance phase (12 days, Study 1): Mean number of normokalaemic days:
| Maintenance phase treatment (once daily) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Placebo | Lokelma | P-value vs. placebo | ||||||||
| Correction phase Lokelma dose | n | Days | n | Days | 1.25 g three times daily | 41 | 7.6 | 49 | 7.2 | NS |
| 2.5 g three times daily | 46 | 6.2 | 54 | 8.6 | 0.008 | |||||
| 5 g three times daily | 68 | 6.0 | 64 | 9.0 | 0.001 | |||||
| 10 g three times daily | 61 | 8.2 | 63 | 10.2 | 0.005 | |||||
NS: not significant
At the end of the maintenance period, when Lokelma was no longer administered, average potassium levels increased to near baseline levels.
Study 2
A multi-phase, placebo-controlled maintenance study with an additional open-label phase
In the correction phase of the study, 258 patients with hyperkalaemia (baseline average 5.6, range 4.1 - 7.2 mmol/L) received 10 g of Lokelma administered three times daily for 48 hours. Reductions in potassium were observed 1 hour after the first 10 g dose of Lokelma. Median time to normokalaemia was 2.2 hours with 66% of patients achieving normokalaemia at 24 hours and 88% at 48 hours. Responses were larger in patients with more severe hyperkalaemia; serum potassium fell 0.8, 1.2 and 1.5 mmol/L in patients with baseline serum potassium <5.5, 5.5-5.9 and ≥6 mmol/L, respectively.
Patients who achieved normokalaemia (potassium levels between 3.5 and 5 mmol/L) were randomised in a double-blind fashion to one of three doses of Lokelma [5 g (n=45), 10 g (n=51), or 15 g (n=56)] or placebo (n=85) administered once daily for 28 days (the double-blind randomised withdrawal phase).
The proportion of subjects with average serum potassium < 5.1 mmol/L from Study Day 8 to 29 (three-week period) was greater at the 5 g, 10 g and 15 g once daily doses of Lokelma (80%, 90% and 94%, respectively), compared with placebo (46%). There was a mean decrease in serum potassium of 0.77 mmol/L, 1.10 mmol/L, 1.19 mmol/L and 0.44 mmol/L, respectively, and the proportion of subjects who remained normokalaemic was 71%, 76%, 85% and 48% in the 5 g, 10 g, 15 g once daily doses of Lokelma and placebo groups, respectively.
Maintenance phase with Lokelma titration (open-label) results: 123 patients entered the 11-month open-label phase. The proportion of subjects with average serum potassium <5.1 mmol/L was 88%, the average serum potassium level was 4.66 mmol/L and the proportion of serum potassium measurements below 3.5 mmol/L was less than 1%; between 3.5 and 5.1 mmol/L was 77%; or between 3.5 and 5.5 mmol/L was 93%, irrespective of other factors that might influence the serum potassium. Treatment was discontinued on study exit (Day 365).
Kaplan-Meier estimates of time to relapse for maintenance phase showed dose dependence in time to relapse with median time for 5 g dose ranging from 4 to 21 days depending on the baseline serum potassium values. Serum potassium should be monitored periodically and the Lokelma dose titrated as described in section 4.2.
Figure 1 illustrates the mean serum potassium over the correction and maintenance phases of the study.
Figure 1. Correction and maintenance phases (Study 2): mean serum potassium over time with 95% CI:
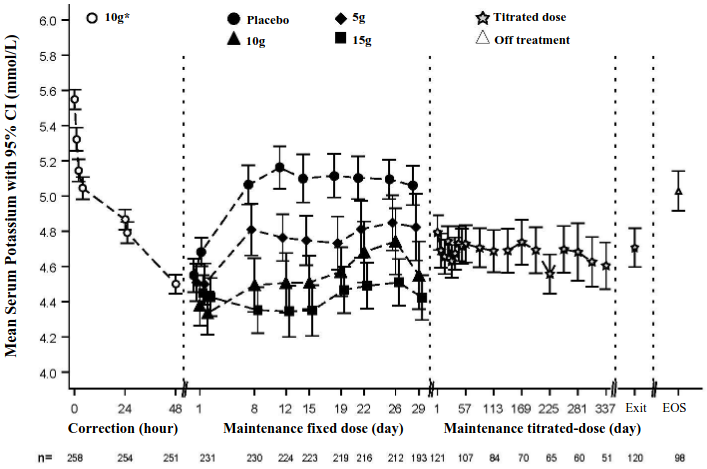
Exit=Last Visit within 1 day of Last Dose, EOS=End of Study (7 days +/- 1 day after Last Dose)
* Given three times daily
Study 3
A study in chronic kidney disease patients with hyperkalaemia
This study was a double-blind placebo-controlled dose-escalating study in 90 patients (60 Lokelma patients; 30 controls) with baseline eGFR between 30-60 ml/min/1.73 m² and hyperkalaemia (baseline serum potassium 5.2 mmol/L, range 4.6-6 mmol/L). Patients were randomised to receive escalating doses of Lokelma (0.3 g, 3 g and 10 g) or placebo, administered three times a day with meals for two to four days. The primary endpoint was the rate of change in serum potassium from baseline throughout the initial 2 days of treatment. The trial met the primary efficacy endpoint at the 3 g and 10 g doses of Lokelma compared to placebo. Lokelma at the 10 g dose and the 3 g dose resulted in mean maximal reductions of 0.92 mmol/L and 0.43 mmol/L, respectively. Twenty-four hour urine collections showed that Lokelma decreased urinary potassium excretion from baseline by 15.8 mmol/24 h compared to placebo increase by 8.9 mmol/24 h (p<0.001). Sodium excretion was unchanged relative to placebo (10 g, increase by 25.4 mmol/24 h compared to placebo increase by 36.9 mmol/24 h (NS)).
Study 4
A two-phase, multicenter, multi-dose, open-label safety and efficacy study
The long term (up to 12 months) effects of Lokelma were assessed in this study in 751 subjects with hyperkalaemia (baseline average 5.59 mmol/L; range 4.3-7.6 mmol/L). Comorbid conditions included chronic kidney disease (65%), diabetes mellitus (64%), heart failure (15%) and hypertension (83%). Use of diuretics and RAAS inhibitors was reported by 51 and 70% of subjects, respectively. During the correction phase, 10 g of Lokelma was administered three times daily for at least 24 hours and up to 72 hours. Subjects who achieved normokalaemia (3.5-5.0 mmol/L, inclusive) within 72 hours entered the maintenance phase of the study. All subjects in the maintenance phase received Lokelma at a starting dose of 5 g once daily which could be increased in increments of 5 g once daily (to a maximum of 15 g once daily) or decreased (to a minimum of 5 g once every other day) based upon the titration regimen.
Normokalaemia was achieved in 494/748 (66%), 563/748 (75%) and 583/748 (78%) of subjects after 24, 48 and 72 hours of correction phase dosing with an average reduction in serum potassium of 0.81 mmol/L, 1.02 mmol/L and 1.10 mmol/L at 24 (n=748), 48 (n=104) and 72 (n=28) hours, respectively. Normokalaemia was dependent on baseline potassium concentration, with subjects with the highest baseline serum potassium concentrations having the most prominent decrease after starting the study drug but with the lowest proportion of subjects achieving normokalaemia. One hundred and twenty-six patients had a baseline serum potassium ≥6.0 mmol/L (mean baseline potassium 6.28 mmol/L). These subjects had a mean reduction of 1.37 mmol/L at the end of the correction phase.
Table 4. Correction phase (Study 4): proportion of subjects with serum potassium concentrations between 3.5 and 5.0 mmol/L, inclusive, or between 3.5 and 5.5 mmol/L, inclusive, by correction phase study day - ITT population:
| Lokelma 10 g three times daily (N=749) | |||||||
|---|---|---|---|---|---|---|---|
| Correction Phase (CP) | Serum potassium 3.5 to 5.0 mmol/L, inclusive | Serum potassium 3.5 to 5.5 mmol/L, inclusive | |||||
| n/N | Proportion | 95% CI | n/N | Proportion | 95% CI | ||
| CP at 24 hours | 494/748 | 0.660 | 0.625, 0.694 | 692/748 | 0.925 | 0.904, 0.943 | |
| CP at 48 hours | 563/748 | 0.753 | 0.720, 0.783 | 732/748 | 0.979 | 0.965, 0.988 | |
| CP at 72 hours/CP Last | 583/748 | 0.779 | 0.748, 0.809 | 738/748 | 0.987 | 0.976, 0.994 | |
Note: One subject had a post-dose value that was more than 1 day after last dose. Therefore, the subject was eligible for the Correction Phase ITT Population; however, the time point was excluded from the analysis.
Normokalaemia was maintained while patients remained on drug and the mean serum potassium increased following discontinuation. Among those patients using RAAS inhibitors at baseline, 89% did not discontinue RAAS inhibitor therapy, 74% were able to maintain the same dose during the maintenance phase and among those not on RAAS inhibitors at baseline, 14% were able to initiate this therapy. During maintenance phase, 75.6% of subjects maintained normokalaemia, despite use of RAAS inhibitors.
Figure 2 illustrates the mean serum potassium over the correction and maintenance phases of the study.
Figure 2. Correction and maintenance phases in 12-month open-label study (Study 4) - mean serum potassium over time with 95% CI:
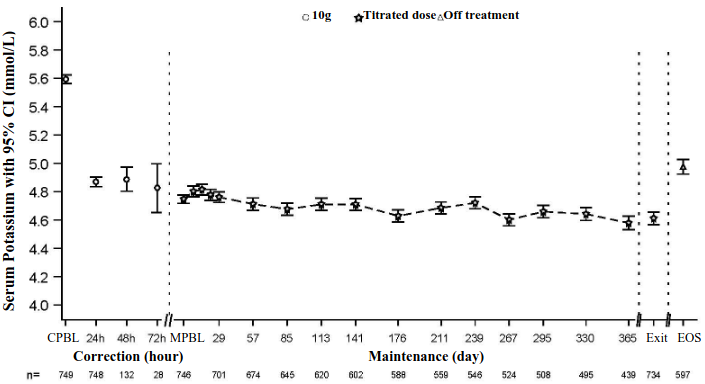
CPBL = Correction Phase Baseline, MPBL = Maintenance Phase Baseline
Exit = Last Visit within 1 day of Last Dose, EOS = End of Study (7 days +/- 1 day after Last Dose)
Study 5
A randomised, double-blind, placebo-controlled study in patients on chronic haemodialysis
In this study, 196 patients (mean age 58 years, range 20 to 86 years) with end stage renal disease on stable dialysis for at least 3 months and persistent pre-dialysis hyperkalaemia were randomised to receive Lokelma 5 g or placebo once daily on non-dialysis days. At randomization, mean serum potassium levels were 5.8 mmol/L (range 4.2-7.3 mmol/L) in the Lokelma group and 5.9 mmol/L (range 4.2–7.3 mmol/L) in the placebo group. To achieve pre-dialysis serum potassium level between 4.0-5.0 mmol/L during the dose adjustment period (initial 4 weeks), the dose could be adjusted weekly in 5 g increments up to 15 g once daily based on pre-dialysis serum potassium measurement after the LIDI. The dose reached at the end of the dose-adjustment period was maintained throughout the subsequent 4-week evaluation period. At the end of the dose adjustment period, 37%, 43%, and 19% of patients were on Lokelma 5 g, 10 g and 15 g. The proportion of responders, defined as those subjects who maintained a pre-dialysis serum potassium between 4.0 and 5.0 mmol/L on at least 3 out of 4 dialysis treatments after LIDI and who did not receive rescue therapy during the evaluation period, was 41% in the Lokelma group, and 1% in the placebo group (p<0.001) (see Figure 3).
In post-hoc analyses the number of times patients had serum potassium between 4.0 and 5.0 mmol/L after the LIDI during the evaluation period was higher in the Lokelma group. 24% of patients were within this range at all 4 visits in the Lokelma group and none in the placebo group. The post-hoc analysis showed the proportion of patients who maintained serum potassium level between 3.5 and 5.5 mmol/L on at least 3 out of 4 dialysis treatments after LIDI during the evaluation period was 70% in the Lokelma group and 21% in the placebo group.
At the end of treatment, the mean post-dialysis serum potassium level was 3.6 mmol/L (range 2.6-5.7 mmol/L) in Lokelma group and 3.9 mmol/L (range 2.2-7.3 mmol/L) in the placebo group. There were no differences between Lokelma and placebo groups in interdialytic weight gain (IDWG). IDWG was defined as pre-dialysis weight minus post-dialysis weight on the previous dialysis session and was measured after the LIDI.
Figure 3. Mean pre-dialysis serum potassium levels over time in patients on chronic dialysis:
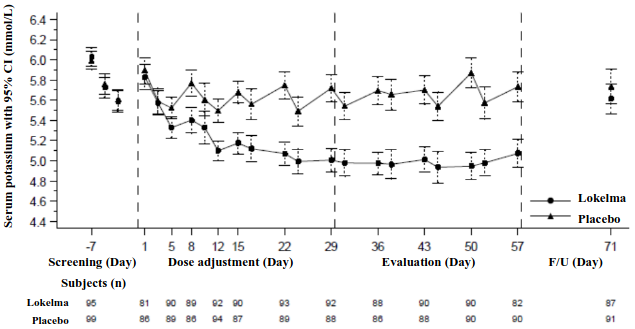
F/U - follow-up period
The displayed error bars correspond to 95% confidence intervals.
n = Number of patients with non-missing potassium measurements at a particular visit.
Study 6 - PRIORITIZE HF
This was a randomised, double-blind, placebo-controlled study aimed to assess if a treatment regimen containing Lokelma would allow Renin Angiotensin Aldosterone System Inhibitor (RAASi) therapies to be up-titrated to target doses at 3 months vs placebo in patients with heart failure and hyperkalaemia or at high risk of developing hyperkalaemia. The primary endpoint of the study was proportion of subjects in the following 4 categories at 3 months: No Angiotensin Converting Enzyme Inhibitors (ACEi)/Angiotensin Receptor Blocker (ARB)/Angiotensin Receptor Blocker/Neprilysin Inhibitors (ARNI) or at less than target dose and no Mineralocorticoid Receptor Antagonist (MRA); ACEi/ARB/ARNI at target dose and no MRA; MRA at less than target dose; MRA at target dose.
Heart failure patients with New York Heart Association (NYHA) Class II-IV with Left Ventricular Ejection Fraction (LVEF) ≤40%, estimated glomerular filtration rate (eGFR) 20-59 mL/min/1.73 m² and serum potassium 4.0-5.5mmol/L were randomised to receive Lokelma or placebo (1:1) for 3 months. RAASi up-titration to guideline-recommended doses was encouraged but not mandated, and Lokelma or placebo dose titrations were performed in parallel to prevent hyperkalaemia.
The study was terminated prematurely during the Covid-19 pandemic due to recruitment challenges and difficulties to ascertain adequate safety monitoring when patients were not able to attend study and laboratory check visits. This resulted in 182 patients randomised as opposed to the planned 280 patients. The premature termination of the study precludes any firm conclusions on the primary and other efficacy measures.
Study 7 - REALIZE-K
This was a Phase 4, prospective, double-blind, randomised-withdrawal trial aimed to determine the efficacy and safety of Lokelma in optimising MRA therapy in patients with heart failure with reduced ejection fraction. The primary endpoint was the occurrence of optimal response, defined as the composite of serum potassium in the normal range (3.5-5.0 mmol/L), on a spironolactone dose of ≥25 mg/daily, without the need of rescue therapy for hyperkalaemia.
This study enrolled adults with established heart failure diagnosis ≥3 months duration, LVEF ≤40% with NYHA Class II-IV symptoms who were receiving treatment with an ACEi/ARB/ARNI and a beta-adrenergic receptor blocker (unless contraindicated) at stable dose for ≥4 weeks. Participation was permitted for those untreated with a MRA and those receiving spironolactone or eplerenone <25 mg once daily.
Patients were screened and entered an open label run in phase with two cohorts. Cohort 1 included patients who had evidence of prevalent hyperkalaemia (defined as serum potassium 5.1-5.9 mmol/L) and eGFR ≥30 mL/min/1.73 m². Patients in this cohort received Lokelma to correct potassium to normal range, after which spironolactone was initiated or up-titrated per protocol. Cohort 2 included patients who were at high risk for hyperkalaemia (defined as either a history of serum potassium >5.0 mmol/L within the prior 36 month and eGFR ≥30 mL/min/1.73 m² OR serum potassium 4.5-5.0 mmol/L with eGFR 30-60 mL/min/1.73 m² OR serum potassium 4.5-5.0 mmol/L and age >75 years. These patients had spironolactone initiated or up-titrated towards the target dose; those developing hyperkalaemia received Lokelma to correct potassium to normal range, while those who failed to become hyperkalaemic within 4 weeks were discontinued from the study.
In this trial, use of Lokelma led to a greater occurrence of optimal response for the primary endpoint compared with placebo (OR 4.45 [95% CI 2.89-6.86], p<0.001, estimated percentages 71% vs 36%). These results were consistent when patients receiving 15 g of Lokelma at randomisation were excluded from the analysis. Lokelma also improved secondary endpoints vs placebo: the occurrence of normokalaemia on the randomised dose of spironolactone and without rescue therapy for hyperkalaemia (HK) (OR 4.58 [95% CI 2.78-7.55], p<0.001; estimated percentages 58% vs 23%); the occurrence of spironolactone ≥25 mg/daily dose (OR 4.33 [95% CI 2.50-7.52], p<0.001; estimated percentages 81% vs 50%); time to first HK episode (serum K+ >5.0 mmol/L) (HR 0.51 [95% CI 0.37- 0.71], p<0.001); and time to first decrease or discontinuation of spironolactone dose due to HK (HR 0.37 [95% CI 0.17-0.73], p=0.006).
Study 8 - STABILIZE-CKD
This was a Phase 3, randomised withdrawal, double-blind, parallel-group, placebo-controlled study aimed to assess whether Lokelma, as an adjunct to ACEi/ARB therapy, is superior to placebo in slowing Chronic kidney disease (CKD) progression over time in patients with hyperkalaemia or at risk of hyperkalaemia. Co-primary endpoints were eGFR total slope (from randomisation to end of treatment) and eGFR chronic slope (from 12 weeks after randomisation to end of treatment).
The study enrolled patients with eGFR 25-59 mL/min/1.73 m², urine albumin-to-creatinine ratio (uACR) 200-5000 mg/g, and hyperkalaemia (serum potassium [sK+] >5.0 to ≤6.5 mmol/L) on adequate/limited ACEi/ARB therapy or normokalaemia on limited ACEi/ARB therapy. Patients with NYHA class III to IV congestive heart failure at the time of screening or previous history of severe or symptomatic heart failure were excluded from the study.
The study included a screening period, an initiation phase (with up to 72 hr open-label Lokelma for the participants to maintain or achieve normokalaemia), a 3-month run-in phase (where lisinopril or valsartan were expected to be up-titrated to maximal tolerated doses under open-label Lokelma potassium management), an originally planned 24-month randomised blinded maintenance phase (1:1 blinded Lokelma or matching placebo, and both lisinopril or valsartan and Lokelma/placebo were titrated and monitored for efficacy and safety assessments), and a follow-up visit.
The trial was terminated early due to recruitment challenges, resulting in a reduced sample size of 760 randomised patients as opposed to the planned 1360 patients, and shortened post-randomisation follow-up duration (median ~8 - 9 months, as opposed to the planned 24 months). This precludes any conclusions on eGFR slope and hard renal outcomes.
In a pooled analysis of placebo-controlled clinical studies of Lokelma in non-dialysis patients (PRIORITIZE-HF, REALIZE-K, STABILIZE-CKD), more patients with pre-existing heart failure experienced worsening of heart failure on Lokelma comparing with the ones on placebo (see section 4.8).
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Lokelma in one or more subsets of the paediatric population, with hyperkalaemia (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Sodium zirconium cyclosilicate is an inorganic, insoluble compound that is not subject to enzymatic metabolism. In addition, clinical trials have shown it not to be systemically absorbed. An in vivo mass balance study in rats showed that sodium zirconium cyclosilicate was recovered in the faeces with no evidence of systemic absorption. Due to these factors and its insolubility, no in vivo or in vitro studies have been performed to examine its effect on cytochrome P450 (CYP450) enzymes or transporter activity.
Elimination
Sodium zirconium cyclosilicate is eliminated via the faeces.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.