MOVANTIK Film-coated tablet Ref.[10882] Active ingredients: Naloxegol
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Naloxegol is an antagonist of opioid binding at the mu-opioid receptor. When administered at the recommended dose levels, naloxegol functions as a peripherally-acting mu-opioid receptor antagonist in tissues, such as the gastrointestinal tract, thereby decreasing the constipating effects of opioids.
Naloxegol is a PEGylated derivative of naloxone and is a substrate for the P-glycoprotein transporter (P-gp). Also, the presence of the PEG moiety in naloxegol reduces its passive permeability as compared with naloxone. Due to the reduced permeability and increased efflux of naloxegol across the blood-brain barrier, related to P-gp substrate properties, the CNS penetration of naloxegol is expected to be negligible at the recommended dose levels limiting the potential for interference with centrally mediated opioid analgesia.
12.2. Pharmacodynamics
Use of opioids induces slowing of gastrointestinal motility and transit. Antagonism of gastrointestinal mu-opioid receptors by naloxegol inhibits opioid-induced delay of gastrointestinal transit time.
Effect on Cardiac Repolarization
In a randomized, double-blind, 4-way cross-over thorough QTc prolongation study with moxifloxacin as a positive control, a single 25 mg therapeutic dose or a 150 mg dose (6 times the maximum recommended dosage) of naloxegol did not have an effect on the QTc interval compared to placebo. Changes in heart rate, RR, PR, and QRS ECG intervals were similar between placebo and naloxegol 25 or 150 mg.
Exposure Response Analysis
The exposure-response analysis for adverse events showed that the probability of experiencing abdominal pain increased with increasing naloxegol exposure over the dose range of 12.5 mg to 25 mg once a day. The exposure-response analysis for efficacy conducted using the definition of response in the clinical trials [see Clinical Studies (14)] indicated that response was similar over this dose range.
12.3. Pharmacokinetics
Absorption
Following oral administration, MOVANTIK is absorbed with peak concentrations (Cmax) achieved at less than 2 hours. In a majority of subjects, a secondary plasma concentration peak of naloxegol was observed approximately 0.4 to 3 hours after the first peak. Across the range of doses evaluated, peak plasma concentration and area under the plasma concentration-time curve (AUC) increased in a dose-proportional or almost dose-proportional manner. Accumulation was minimal following multiple daily doses of naloxegol.
MOVANTIK as a crushed tablet mixed in water, given orally or administered through a nasogastric tube into the stomach, provides systemic naloxegol concentrations that are comparable to the whole tablet, with a median tmax of 0.75 and 1.5 hours (range 0.25 to 5 hours) for the crushed tablet given orally and the crushed tablet given via nasogastric (NG) tube, respectively [see Dosage and Administration (2.2)].
Food Effects
A high-fat meal increased the extent and rate of naloxegol absorption. The Cmax and AUC were increased by approximately 30% and 45%, respectively. In clinical trials, naloxegol was dosed on an empty stomach approximately 1 hour prior to the first meal in the morning.
Distribution
The mean apparent volume of distribution during the terminal phase (Vz/F) in healthy volunteers ranged from 968 L to 2140 L across dosing groups and studies. Plasma protein binding of naloxegol in humans was low (~4.2%).
Metabolism
Naloxegol is metabolized primarily by the CYP3A enzyme system. In a mass balance study in humans, a total of 6 metabolites were identified in plasma, urine, and feces. These metabolites were formed via N-dealkylation, O-demethylation, oxidation, and partial loss of the PEG chain. Human metabolism data suggests absence of major metabolites. The activity of the metabolites at the opioid receptor has not been determined.
Excretion
Following oral administration of radio-labeled naloxegol, 68% and 16% of total administered dose were recovered in the feces and urine, respectively. Parent naloxegol excreted in the urine accounted for less than 6% of the total administered dose. Approximately 16% of radioactivity in feces was noted to be unchanged naloxegol, while the remaining was attributed to metabolites. Thus, renal excretion is a minor clearance pathway for naloxegol. In a clinical pharmacology study, the half-life of naloxegol at therapeutic doses ranged from 6 to 11 hours.
Specific Populations
Renal Impairment
The effect of renal impairment on the pharmacokinetics of a 25 mg single oral dose of MOVANTIK was studied in subjects with renal impairment (RI) classified as moderate (n=8), severe (n=4), or end-stage renal disease (ESRD) not yet on dialysis (n=4), and compared with healthy subjects (n=6). Most renal impairment (RI) subjects (6 out of 8 with moderate RI, 3 out of 4 with severe RI, and 3 out of 4 with ESRD) had plasma naloxegol pharmacokinetics comparable to those in healthy subjects. The remaining individuals with renal impairment demonstrated higher naloxegol exposures (up to 10-fold) compared to the control group. The reason for these high exposures is unknown.
This study also included 8 ESRD patients on hemodialysis. Plasma concentrations of naloxegol in these subjects were similar to healthy volunteers with normal renal function, when MOVANTIK was administered either pre- or post-hemodialysis [see Dosage and Administration (2.3), Use in Specific Populations (8.6), and Overdosage (10)].
Hepatic Impairment
Slight decreases in AUC of naloxegol were observed in subjects with mild and moderate hepatic impairment (Child-Pugh Classes A and B; n=8 per group) compared to subjects with normal hepatic function (n=8), following administration of a single 25 mg oral dose of MOVANTIK. The effect of severe hepatic impairment (Child-Pugh Class C) on the pharmacokinetics of naloxegol was not evaluated [see Use in Specific Populations (8.7)].
Age:
The mean Cmax,ss and AUCτ,ss values seen in elderly healthy Japanese subjects (n=6) were approximately 45% and 54% greater than those obtained in young healthy subjects (n=6) following multiple daily doses of naloxegol (25 mg).
Gender
There is no gender effect on the pharmacokinetics of naloxegol.
Race
When compared to Caucasian subjects, naloxegol AUC was approximately 20% lower in Blacks and Cmax was approximately 10% lower and 30% higher in Blacks and Asians, respectively.
Drug Interaction Studies
Effect of MOVANTIK on Other Drugs
In in vitro studies at clinically relevant concentrations, naloxegol did not show a significant inhibitory effect on the activity of CYP1A2, CYP2C8, CYP2C9, CYP2D6, CYP3A4 or CYP2C19, nor a significant induction effect on the activity of CYP1A2, CYP2B6, or CYP3A4. Therefore, MOVANTIK is not expected to alter the metabolic clearance of co-administered drugs that are metabolized by these enzymes. Naloxegol is not a significant inhibitor of P-gp, BCRP, OAT1, OAT3, OCT2, OATP1B1, and OATP1B3.
In healthy subjects receiving morphine 5 mg/70 kg intravenously, single doses of MOVANTIK ranging from 8 mg to 1000 mg were given concomitantly with 5 to 6 subjects per dose cohort. With increasing MOVANTIK dose, there was no increasing or decreasing trend in morphine exposure compared to morphine administered alone. An analysis of the pooled data indicated that MOVANTIK had no meaningful impact on the systemic exposure of morphine and its major circulating metabolites.
Effect of Other Drugs on MOVANTIK
Naloxegol is metabolized mainly by CYP3A enzymes and is a substrate of P-gp transporter. The effects of co-administered drugs on the pharmacokinetics of naloxegol are summarized in Figure 1 [see Drug Interactions (7.1)].
The effects of once daily oral dosing of 400 mg ketoconazole, once daily oral dosing of 600 mg rifampicin and once daily oral dosing of 240 mg diltiazem (as an extended release formulation) on the pharmacokinetics of 25 mg MOVANTIK were studied following multiple dosing and at steady state exposure of the perpetrator drugs. The effects of 600 mg oral dosing of quinidine and intravenous morphine (5 mg/70 kg) on the pharmacokinetics of 25 mg MOVANTIK were studied following single dosing of the perpetrator drugs.
Figure 1. Effect of Co-administered Drugs on the Pharmacokinetics of Naloxegol:
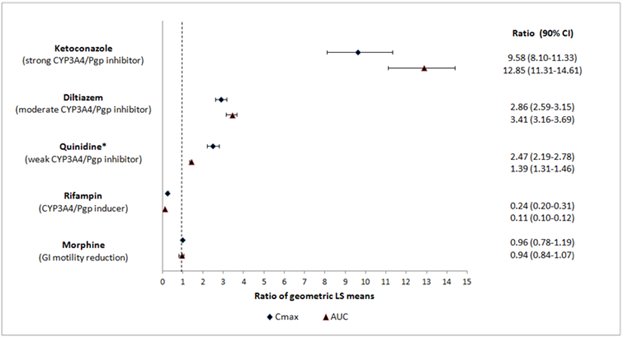
* Quinidine due to its effect on P-gp transporter increased naloxegol Cmax by 2.5-fold; the AUC increased by 1.4-fold; no dosage adjustment is necessary.
No drug interaction studies have been conducted for MOVANTIK with drugs that alter gastric pH (e.g., antacids, proton-pump inhibitors).
Simulations using physiologically based pharmacokinetic modeling, suggested that naloxegol exposures after co-administration of a single oral 25 mg dose of MOVANTIK with a moderate CYP3A inducer efavirenz (400 mg once a day) are similar to those after 12.5 mg MOVANTIK alone.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In a 104-week carcinogenicity study in CD-1 mice, naloxegol was not tumorigenic at oral doses up to 100 mg/kg/day in males and 160 mg/kg/day in females (43 and 27 times the human AUC at the maximum recommended human dose for male and female mice, respectively). In a carcinogenicity study in Sprague-Dawley rats, naloxegol was administered orally at doses of 40, 120, and 400 mg/kg/day for at least 93 weeks. Naloxegol did not cause an increase in tumors in female rats. In male rats, an increase in interstitial (Leydig) cell adenomas in testes was observed at 400 mg/kg/day (818 times the human AUC at the maximum recommended human dose). The no observed effect level for increased tumor incidence was 120 mg/kg/day in male and 400 mg/kg/day in female rats (246 and 1030 times the human AUC at the maximum recommended human dose for male and female rats, respectively). The Leydig cell neoplasms in rats are considered to be unlikely relevant to humans.
Mutagenesis
Naloxegol was not genotoxic in the in vitro bacterial reverse mutation (Ames) assay, mouse lymphoma TK +/- mutation assay, or the in vivo mouse micronucleus assay.
Impairment of Fertility
Naloxegol was found to have no effect on fertility or reproductive performance in male and female rats at oral doses up to 1000 mg/kg/day (greater than 1000 times the human AUC at the maximum recommended human dose).
14. Clinical Studies
The safety and efficacy of MOVANTIK were evaluated in two replicate, randomized, double-blind placebo-controlled trials (Study 1 and Study 2) in patients with opioid-induced constipation (OIC) and non-cancer related pain.
Patients receiving an opioid morphine equivalent daily dose of between 30 mg and 1,000 mg for at least four weeks before enrollment and self-reported OIC were eligible to participate. OIC was confirmed through a two-week run-in period and was defined as <3 spontaneous bowel movements (SBMs) per week on average with at least 25% of the SBMs associated with one or more of the following conditions: (1) straining, (2) hard or lumpy stools; and (3) having a sensation of incomplete evacuation. An SBM was defined as a bowel movement (BM) without rescue laxative taken within the past 24 hours. Patients with 0 BMs over the two-week run-in period or patients with an uneven distribution of SBMs across the two-week run-in period (0 SBMs in one week with ≥4 SBMs in the other week) were excluded. Throughout the studies (including the two-week run-in period), patients were prohibited from using laxatives other than bisacodyl rescue laxative (if they had not had a BM for 72 hours) and one-time use of an enema (if after 3 doses of bisacodyl, they still did not have a BM).
Patients suspected of having clinically important disruptions to the blood-brain barrier were not enrolled in these studies.
A total of 652 patients in Study 1 and 700 patients in Study 2 were randomized in a 1:1:1 ratio to receive 12.5 mg or 25 mg of MOVANTIK or placebo once daily for 12 weeks.
The mean age of the subjects in these two studies was 52 years, 10% and 13% were 65 years of age or older, 61% and 63% were women, and 78% and 80% were White in Studies 1 and 2, respectively.
Back pain was the most common reason for pain (56% and 57%); arthritis (10% and 10%) and joint pain (3% and 5%) were other prominent reasons in Studies 1 and 2, respectively. Prior to enrollment, patients had been using their current opioid for an average of 3.6 and 3.7 years. The patients who participated in Studies 1 and 2 were taking a wide range of opioids. The mean baseline opioid morphine equivalent daily dosage was 140 mg and 136 mg per day.
Use of one or more laxatives on at least one occasion within the two weeks prior to enrollment was reported by 71% of patients in both Studies 1 and 2.
The primary endpoint was response defined as: ≥3 SBMs per week and a change from baseline of ≥1 SBM per week for at least 9 out of the 12 study weeks and 3 out of the last 4 weeks.
There was a statistically significant difference for the 25 mg MOVANTIK treatment group versus placebo for the primary endpoint in Study 1 and Study 2 (see Table 3). Statistical significance for the 12.5 mg treatment group versus placebo was observed in Study 1 but not in Study 2 (see Table 3).
Table 3. Primary Endpoint: Response* (Studies 1 and 2):
| Study 1 | |||
|---|---|---|---|
| Placebo (N=214) | 12.5 mg (N=213) | 25 mg (N=214) | |
| Patients responding, n (%) | 63 (29%) | 87 (41%) | 95 (44%) |
| Treatment Difference (MOVANTIK-Placebo) | -- | 11.4% | 15.0% |
| 95% Confidence Interval | -- | (2.4%, 20.4%) | (5.9%, 24.0%) |
| p-value | -- | 0.015† | 0.001† |
| Study 2 | |||
| Placebo (N=232) | 12.5 mg (N=232) | 25 mg (N=232) | |
| Patients responding, n (%) | 68 (29%) | 81 (35%) | 92 (40%) |
| Treatment Difference (MOVANTIK-Placebo) | -- | 5.6% | 10.3% |
| 95% Confidence Interval | -- | (-2.9%, 14.1%) | (1.7%, 18.9%) |
| p-value | -- | 0.202 | 0.021† |
* Response defined as: ≥3 SBMs per week and change from baseline of ≥1 SBM per week for at least 9 out of the 12 study weeks and 3 out of the last 4 weeks.
† Statistically significant: p-values based on the Cochran-Mantel-Haenszel test.
One secondary endpoint in Study 1 and Study 2 was response in laxative users with OIC symptoms. This subgroup comprised 55% and 53% of total patients in these two studies, respectively. These patients (identified using an investigator-administered questionnaire), prior to enrollment, had reported using laxative(s) at least 4 out of the past 14 days with at least one of the following OIC symptoms of moderate, severe, or very severe intensity: incomplete bowel movements, hard stool, straining, or sensation of needing to pass a bowel movement but unable to do so. In this subgroup, in Studies 1 and 2, 42% and 50% reported using laxatives on a daily basis. The most frequently reported laxatives used on a daily basis were stool softeners (18% and 24%), stimulants (16% and 18%), and polyethylene glycol (6% and 5%). Use of two laxative classes was reported in 31% and 27% anytime during the 14 days prior to enrollment. The most commonly reported combination was stimulants and stool softeners (10% and 8%). In Study 1, a statistically significantly higher percentage of patients in this subgroup responded with MOVANTIK 12.5 mg compared to placebo (43% vs. 29%; p=0.03) and with MOVANTIK 25 mg compared to placebo (49% vs. 29%; p=0.002). In Study 2, a statistically significantly higher percentage of patients in this subgroup responded with MOVANTIK 25 mg compared to placebo (47% vs. 31%; p=0.01). This secondary endpoint was not tested for MOVANTIK 12.5 mg versus placebo in Study 2 because the primary endpoint was not statistically significant.
Another secondary endpoint was time to first post-dose SBM. The time to first post-dose SBM was significantly shorter with MOVANTIK 25 mg compared to placebo in both Study 1 (p<0.001) and Study 2 (p<0.001), and for MOVANTIK 12.5 mg as compared to placebo in Study 1 (p<0.001). For Study 1, the median times to first post-dose SBM were 6, 20, and 36 hours with MOVANTIK 25 mg, MOVANTIK 12.5 mg, and placebo, respectively. For Study 2, the median times to first post-dose SBM were 12 and 37 hours with MOVANTIK 25 mg and placebo, respectively. These analyses do not include the results for MOVANTIK 12.5 mg versus placebo in Study 2 because the primary endpoint was not statistically significant. In the two studies, 61-70% and 58% of patients receiving MOVANTIK 25 mg and MOVANTIK 12.5 mg, respectively, had an SBM within 24 hours of the first dose.
A third secondary endpoint was an evaluation of change from baseline between the treatment groups for mean number of days per week with at least 1 SBM but no more than 3 SBMs. There was a significant difference in number of days per week with 1 to 3 SBMs per day on average over 12 weeks between MOVANTIK 25 mg (Study 1 and Study 2) and MOVANTIK 12.5 mg (Study 1) and placebo.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.