NUPLAZID Capsule Ref.[10166] Active ingredients: Pimavanserin
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
The mechanism of action of pimavanserin in the treatment of hallucinations and delusions associated with Parkinson's disease psychosis is unclear. However, the effect of pimavanserin could be mediated through a combination of inverse agonist and antagonist activity at serotonin 5-HT2A receptors and to a lesser extent at serotonin 5-HT2C receptors.
12.2. Pharmacodynamics
In vitro, pimavanserin acts as an inverse agonist and antagonist at serotonin 5-HT2A receptors with high binding affinity (Ki value 0.087 nM) and at serotonin 5-HT2C receptors with lower binding affinity (Ki value 0.44 nM). Pimavanserin shows low binding to sigma 1 receptors (Ki value 120 nM) and has no appreciable affinity (Ki value >300 nM), to serotonin 5-HT2B, dopaminergic (including D2), muscarinic, histaminergic, or adrenergic receptors, or to calcium channels.
Cardiac Electrophysiology
The effect of NUPLAZID on the QTc interval was evaluated in a randomized placebo- and positive-controlled double-blind, multiple-dose parallel thorough QTc study in 252 healthy subjects. A central tendency analysis of the QTc data at steady-state demonstrated that the maximum mean change from baseline (upper bound of the two-sided 90% CI) was 13.5 (16.6) msec at a dose of twice the therapeutic dose. A pharmacokinetic/pharmacodynamic analysis with NUPLAZID suggested a concentration-dependent QTc interval prolongation in the therapeutic range.
In the 6-week, placebo-controlled effectiveness studies, mean increases in QTc interval of ~5-8 msec were observed in patients receiving once-daily doses of NUPLAZID 34 mg. These data are consistent with the profile observed in a thorough QT study in healthy subjects. Sporadic QTcF values ≥500 msec and change from baseline values ≥60 msec were observed in subjects treated with NUPLAZID 34 mg; although the incidence was generally similar for NUPLAZID and placebo groups. There were no reports of torsade de pointes or any differences from placebo in the incidence of other adverse reactions associated with delayed ventricular repolarization in studies of NUPLAZID, including those patients with hallucinations and delusions associated with PDP [see Warnings and Precautions (5.2)].
12.3. Pharmacokinetics
Pimavanserin demonstrates dose-proportional pharmacokinetics after single oral doses from 17 to 255 mg (0.5- to 7.5-times the recommended dosage). The pharmacokinetics of pimavanserin are similar in both the study population and healthy subjects. The mean plasma half-lives for pimavanserin and the active metabolite (N-desmethylated metabolite) are approximately 57 hours and 200 hours, respectively.
Absorption
The median Tmax of pimavanserin was 6 (range 4-24) hours and was generally unaffected by dose. The bioavailability of pimavanserin oral tablet and pimavanserin solution was essentially identical. The formation of the major circulating N-desmethylated metabolite AC-279 (active) from pimavanserin occurs with a median Tmax of 6 hours.
Administration of one 34 mg capsule once daily results in plasma pimavanserin concentrations that are similar to exposure with two 17 mg tablets once daily.
Effect of Food
Ingestion of a high-fat meal had no significant effect on rate (Cmax) and extent (AUC) of pimavanserin exposure. Cmax decreased by about 9% while AUC increased by about 8% with a high-fat meal.
Distribution
Pimavanserin is highly protein bound (~95%) in human plasma. Protein binding appeared to be dose-independent and did not change significantly over dosing time from Day 1 to Day 14. Following administration of a single dose of NUPLAZID (34 mg), the mean (SD) apparent volume of distribution was 2173 (307) L.
Elimination
Metabolism
Pimavanserin is predominantly metabolized by CYP3A4 and CYP3A5 and to a lesser extent by CYP2J2, CYP2D6, and various other CYP and FMO enzymes. CYP3A4 is the major enzyme responsible for the formation of its major active metabolite (AC-279). Pimavanserin does not cause clinically significant CYP inhibition or induction of CYP3A4. Based on in vitro data, pimavanserin is not an irreversible inhibitor of any of the major hepatic and intestinal human CYP enzymes involved in drug metabolism (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4).
Based on in vitro studies, transporters play no significant role in the disposition of pimavanserin.
AC-279 is neither a reversible or irreversible (metabolism-dependent) inhibitor of any of the major hepatic and intestinal human CYP enzymes involved in drug metabolism (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4). AC-279 does not cause clinically significant CYP3A induction and is not predicted to cause induction of any other CYP enzymes involved in drug metabolism.
Excretion
Approximately 0.55% of the 34 mg oral dose of 14C-pimavanserin was eliminated as unchanged drug in urine and 1.53% was eliminated in feces after 10 days.
Less than 1% of the administered dose of pimavanserin and its active metabolite AC-279 were recovered in urine.
Specific Populations
Population PK analysis indicated that age, sex, ethnicity, and weight do not have clinically relevant effect on the pharmacokinetics of pimavanserin. In addition, the analysis indicated that exposure of pimavanserin in patients with mild to moderate renal impairment was similar to exposure in patients with normal renal function.
The effects of other intrinsic factors on pimavanserin pharmacokinetics is shown in Figure 1 [see Use in Specific Populations (8.6, 8.7)].
Figure 1. Effects of Intrinsic Factors on Pimavanserin Pharmacokinetics:
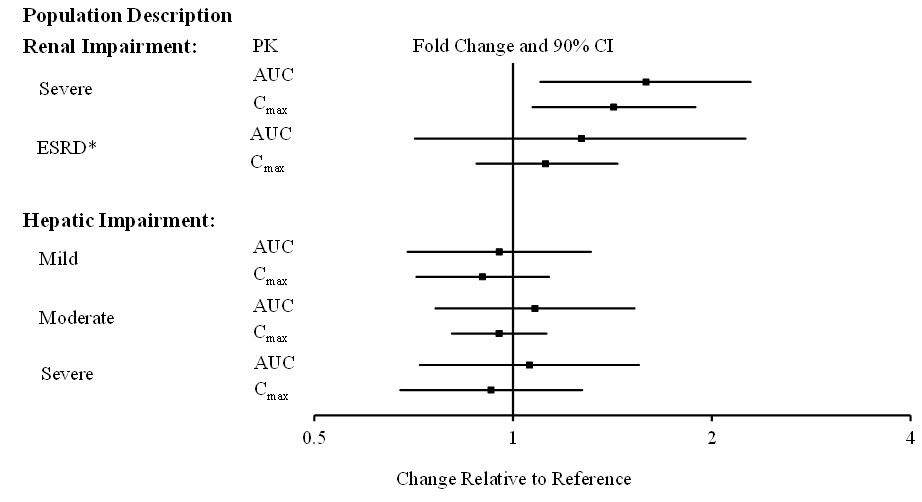
* Less than 10% of the administered dose of NUPLAZID was recovered in the dialysate.
Drug Interaction Studies
CYP3A4 Inhibitor: ketoconazole, a strong inhibitor of CYP3A4, increased pimavanserin Cmax by 1.5-fold and AUC by 3-fold. Population PK modeling and simulation show that steady-state exposure (Cmax,ss and AUCtau) for 10 mg pimavanserin with ketoconazole is similar to exposure for 34 mg pimavanserin alone [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
CYP3A4 Inducer: In a clinical study where single doses of 34 mg pimavanserin were administered on Days 1 and 22, and 600 mg rifampin, a strong inducer of CYP3A4, was given daily on Days 15 through 21, pimavanserin Cmax and AUC decreased by 71% and 91%, respectively, compared to pre-rifampin plasma concentrations. In a simulation with a moderate CYP3A4 inducer (efavirenz), physiologically based pharmacokinetic (PBPK) models predicted pimavanserin Cmax,ss and AUCtau at steady state decreased by approximately 60% and 70%, respectively [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
There is no effect of pimavanserin on the pharmacokinetics of midazolam, a CYP3A4 substrate, or carbidopa/levodopa as shown in Figure 2.
Figure 2. Effects of Pimavanserin on the Pharmacokinetics of Other Drugs:
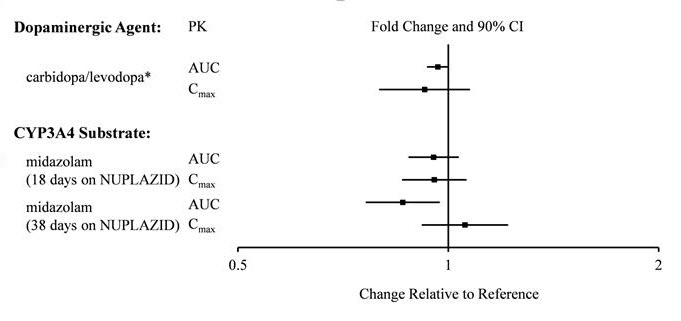
* AUC and Cmax depict levodopa levels.
13.1. Carcinogensis, Mutagenesis, Impairment of Fertility
Carcinogenesis
There was no increase in the incidence of tumors following daily oral administration of pimavanserin to mice or rats for 2 years. Mice were administered pimavanserin at oral doses of 2.6, 6, and 13 (males)/8.5, 21, and 43 mg/kg/day (females) which are 0.01- to 1- (males)/0.5- to 7- (females) times the MRHD of 34 mg/day based on AUC. Rats were administered pimavanserin at oral doses of 2.6, 8.5, and 26 (males)/4.3, 13, and 43 mg/kg/day (females) which are 0.01- to 4- (males)/0.04- to 16- (females) times the MRHD of 34 mg/day based on AUC.
Mutagenesis
Pimavanserin was not mutagenic in the in vitro Ames reverse mutation test, or in the in vitro mouse lymphoma assay, and was not clastogenic in the in vivo mouse bone marrow micronucleus assay.
Impairment of Fertility
Pimavanserin was administered orally to male and female rats before mating, through mating, and up to Day 7 of gestation at doses of 8.5, 51, and 77 mg/kg/day, which are approximately 2-, 15-, and 22-times the maximum recommended human dose (MRHD) of 34 mg/day based on mg/m², respectively. Pimavanserin had no effect on fertility or reproductive performance in male and female rats at doses up to 22-times the MRHD of 34 mg based on mg/m². Changes in uterine parameters (decreases in the number of corpora lutea, number of implants, viable implants, and increases in pre-implantation loss, early resorptions and post-implantation loss) occurred at the highest dose which was also a maternally toxic dose. Changes in sperm parameters (decreased density and motility) and microscopic findings of cytoplasmic vacuolation in the epididymis occurred at doses approximately 15-times the MRHD of 34 mg/day based on mg/m².
13.2. Animal Toxicology and/or Pharmacology
Phospholipidosis (foamy macrophages and/or cytoplasmic vacuolation) was observed in multiple tissues and organs of mice, rats, and monkeys following oral daily administration of pimavanserin. The occurrence of phospholipidosis was both dose- and duration-dependent. The most severely affected organs were the lungs and kidneys. In rats, diffuse phospholipidosis was associated with increased lung and kidney weights, respiratory-related clinical signs including rales, labored breathing, and gasping, renal tubular degeneration, and, in some animals, focal/multifocal chronic inflammation in the lungs at exposures ≥10-times those at the maximum recommended human dose (MRHD) of 34 mg/day based on AUC. Phospholipidosis caused mortality in rats at exposures ≥16-times the MRHD of 34 mg/day based on AUC. The chronic inflammation in the rat lung was characterized by minimal to mild focal collagen positive fibroplasia as shown by specialized staining. Chronic inflammation of the lungs was not seen in monkeys treated for 12 months (exposures 9-times the MRHD). Based on the exposures at the estimated No Observed Effect Level (NOEL) for chronic lung inflammation in rats, there is a 5- to 9-times safety margin after 6-months of treatment and a 2- to 4-times safety margin after 24-months (lifetime) treatment compared to exposure at the MRHD. The relevance of these findings to human risk is not clear.
14. Clinical Studies
The efficacy of NUPLAZID 34 mg as a treatment of hallucinations and delusions associated with Parkinson's disease psychosis was demonstrated in a 6-week, randomized, placebo-controlled, parallel-group study. In this outpatient study, 199 patients were randomized in a 1:1 ratio to NUPLAZID 34 mg or placebo once daily. Study patients (male or female and aged 40 years or older) had a diagnosis of Parkinson's disease (PD) established at least 1 year prior to study entry and had psychotic symptoms (hallucinations and/or delusions) that started after the PD diagnosis and that were severe and frequent enough to warrant treatment with an antipsychotic. At entry, patients were required to have a Mini-Mental State Examination (MMSE) score ≥21 and to be able to self-report symptoms. The majority of patients were on PD medications at entry; these medications were required to be stable for at least 30 days prior to study start and throughout the study period.
The PD-adapted Scale for the Assessment of Positive Symptoms (SAPS-PD) was used to evaluate the efficacy of NUPLAZID 34 mg. SAPS-PD is a 9-item scale adapted for PD from the Hallucinations and Delusions domains of the SAPS. Each item is scored on a scale of 0-5, with 0 being none and 5 representing severe and frequent symptoms. Therefore, the SAPS-PD total score can range from 0 to 45 with higher scores reflecting greater severity of illness. A negative change in score indicates improvement. Primary efficacy was evaluated based on change from baseline to Week 6 in SAPS-PD total score.
As shown in Table 3, Figure 3, and Figure 4, NUPLAZID 34 mg (n=95) was statistically significantly superior to placebo (n=90) in decreasing the frequency and/or severity of hallucinations and delusions in patients with PDP as measured by central, independent, and blinded raters using the SAPS-PD scale. An effect was seen on both the hallucinations and delusions components of the SAPS-PD.
Table 3. Primary Efficacy Analysis Result Based on SAPS-PD (N=185):
| Endpoint | Treatment Group | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Difference* (95% CI) |
|---|---|---|---|---|
| SAPS-PD | NUPLAZID | 15.9 (6.12) | -5.79 (0.66) | -3.06† (-4.91, -1.20) |
| Placebo | 14.7 (5.55) | -2.73 (0.67) | -- | |
| SAPS-PD Hallucinations‡ | NUPLAZID | 11.1 (4.58) | -3.81 (0.46) | -2.01 (-3.29, -0.72) |
| Placebo | 10.0 (3.80) | -1.80 (0.46) | -- | |
| SAPS-PD Delusions‡ | NUPLAZID | 4.8 (3.59) | -1.95 (0.32) | -0.94 (-1.83, -0.04) |
| Placebo | 4.8 (3.82) | -1.01 (0.32) | -- |
SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval.
The effect of NUPLAZID on SAPS-PD improved through the six-week trial period, as shown in Figure 3.
Figure 3. SAPS-PD Change from Baseline through 6 Weeks Total Study Treatment:
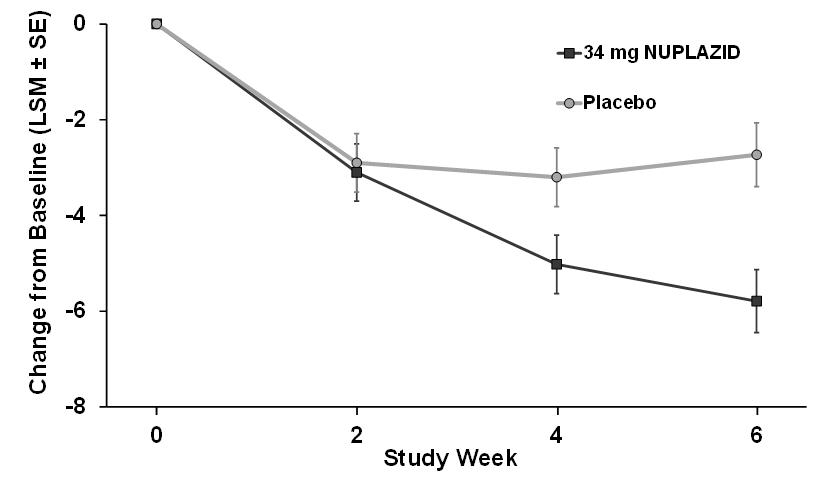
Figure 4. Proportion of Patients with SAPS-PD Score Improvement at the End of Week 6 (N=185):
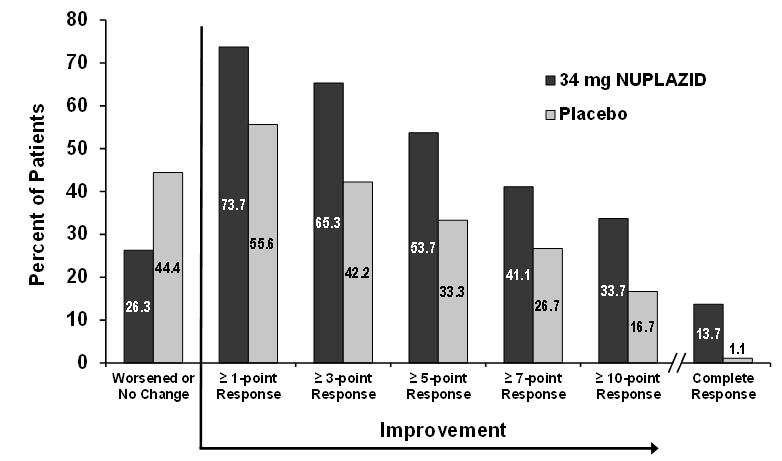
Complete response = SAPS-PD score reduced to zero from baseline value.
Patients with missing values were counted as non-responders.
Motor Function in Patients with Hallucinations and Delusions Associated with Parkinson's Disease Psychosis
NUPLAZID 34 mg did not show an effect compared to placebo on motor function, as measured using the Unified Parkinson's Disease Rating Scale Parts II and III (UPDRS Parts II+III) (Figure 5). A negative change in score indicates improvement. The UPDRS Parts II+III was used to assess the patient's Parkinson's disease state during the 6-week double-blind treatment period. The UPDRS score was calculated as the sum of the 40 items from activities of daily living and motor examination, with a range of 0 to 160.
Figure 5. Motor Function Change from Baseline to Week 6 in UPDRS Parts II+III (LSM - SE):
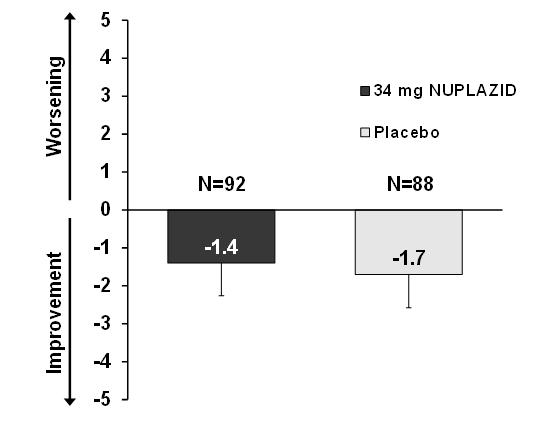
LSM: least-squares mean; SE: standard error. The error bars extend one SE below the LSM.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.