OPDIVO Solution for injection Ref.[116349] Active ingredients:
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Bristol-Myers Squibb Pharma EEIG, Plaza 254, Blanchardstown Corporate Park 2, Dublin 15, D15 T867, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, monoclonal antibodies and antibody drug conjugates, PD-1/PDL-1 (Programmed cell death protein 1/death ligand 1) inhibitors
ATC code: L01FF01
OPDIVO solution for injection contains the active substance nivolumab, which provides the therapeutic effect of this medicinal product, and recombinant human hyaluronidase (rHuPH20), an enzyme used to increase the dispersion and absorption of co-formulated substances when administered subcutaneously.
Mechanism of action
Nivolumab is a human immunoglobulin G4 (IgG4) monoclonal antibody (HuMAb), which binds to the programmed death-1 (PD-1) receptor and blocks its interaction with PD-L1 and PD-L2. The PD-1 receptor is a negative regulator of T-cell activity that has been shown to be involved in the control of T-cell immune responses. Engagement of PD-1 with the ligands PD-L1 and PD-L2, which are expressed in antigen presenting cells and may be expressed by tumours or other cells in the tumour microenvironment, results in inhibition of T-cell proliferation and cytokine secretion. Nivolumab potentiates T-cell responses, including anti-tumour responses, through blockade of PD-1 binding to PD-L1 and PD-L2 ligands. In syngeneic mouse models, blocking PD-1 activity resulted in decreased tumour growth.
Combined nivolumab (anti-PD-1) and ipilimumab (anti-CTLA-4) mediated inhibition results in improved anti-tumour responses in metastatic melanoma. In murine syngeneic tumour models, dual blockade of PD-1 and CTLA-4 resulted in synergistic anti-tumour activity.
Clinical efficacy and safety
Subcutaneous formulation:
Results from a simulation-based pharmacokinetic bridging analyses showed that across all solid tumour types evaluated, subcutaneous nivolumab dosing regimens (600 mg every 2 weeks and 1200 mg every 4 weeks) produced exposures that were noninferior (geometric mean ratio >1) to those for the approved intravenous nivolumab dosing regimens (240 mg every 2 weeks and 480 mg every 4 weeks). Geometric mean exposures were also below those for intravenous nivolumab 10 mg/kg Q2W, a regimen shown to be safe in clinical studies.
The subcutaneous nivolumab clinical safety profile was comparable to intravenous nivolumab.
Melanoma
Adjuvant treatment of melanoma
Randomised phase 3 study of nivolumab vs. placebo (CA20976K)
The safety and efficacy of nivolumab 480 mg monotherapy for the treatment of patients with completely resected melanoma were evaluated in a phase 3, randomised, double-blind study (CA20976K). The study included patients with an ECOG performance status score of 0 or 1 who had Stage IIB or IIC American Joint Committee on Cancer (AJCC), 8th edition, histologically confirmed melanoma that had been completely surgically resected. Enrolment required complete resection of the primary melanoma with negative margins and a negative sentinel lymph node biopsy within 12 weeks prior to randomisation. Patients were enrolled regardless of their tumour PD-L1 status. The study excluded patients with ocular/uveal or mucosal melanoma, active autoimmune disease, any condition requiring systemic treatment with either corticosteroids (≥10 mg daily prednisone or equivalent) or other immunosuppressive medications, as well as patients with prior therapy for melanoma except surgery.
A total of 790 patients were randomised (2:1) to receive either nivolumab (n=526) administered intravenously over 30 minutes at 480 mg every 4 weeks or placebo (n=264) for up to 1 year or until disease recurrence or unacceptable toxicity. Randomisation was stratified by AJCC 8th edition T-category (T3b vs. T4a vs. T4b). Tumour assessments were conducted every 26 weeks during years 1-3 and every 52 weeks from 3 years to 5 years. The primary efficacy outcome measure was recurrence-free survival (RFS). RFS, assessed by the investigator, was defined as the time between the date of randomisation and the date of first recurrence (local, regional, or distant metastasis), new primary melanoma, or death from any cause, whichever occurred first. The secondary outcome measures included OS and distant metastasis-free survival (DMFS).
Baseline characteristics were generally balanced between the two groups. The median age was 62 years (range: 19-92), 61% were men, and 98% were white. Baseline ECOG performance status score was 0 (94%) or 1 (6%). Sixty percent had stage IIB and 40% had stage IIC.
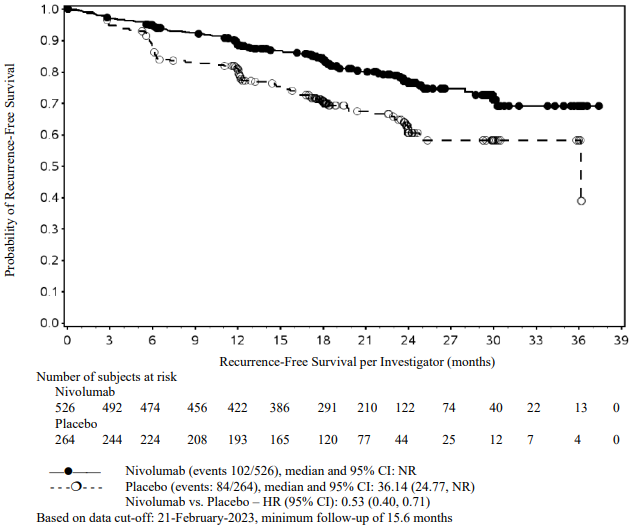
At a primary pre-specified interim analysis (minimum follow-up 7.8 months) a statistically significant improvement in RFS was demonstrated with nivolumab compared to placebo with a HR of 0.42 (95% CI: 0.30, 0.59; p<0.0001). At an updated descriptive RFS analysis (minimum follow-up of 15.6 months), nivolumab continued to demonstrate an RFS improvement with a HR of 0.53 (95% CI: 0.40, 0.71). OS was not mature. At an additional RFS descriptive analysis (minimum follow-up 26.9 months), nivolumab continued to demonstrate an RFS improvement with a HR of 0.62 (95% CI: 0.47-0.80). The median follow-up was 34.25 months for the nivolumab arm and 33.92 months for the placebo arm. The outcomes were consistent with the formal interim analysis. Results reported from the analyses with minimum follow-up of 15.6 months are summarised in Table 14 and Figure 1.
Table 14. Efficacy results (CA20976K):

a Based on stratified Cox proportional hazard model.
b Based on Kaplan-Meier estimates.
RFS benefit was consistent across key subgroups, including disease stage, T-category, and age.
Figure 1. Recurrence-free survival (CA20976K):
Tumour PD-L1 expression data were available for 302/790 (38.2%) randomised patients (36.3% and 42.0% in the nivolumab and placebo arms, respectively), as PD-L1 expression was not a stratification factor for randomisation. The exploratory RFS analyses by PD-L1 expression showed a HR for nivolumab vs placebo of 0.43 (95% CI: 0.22, 0.84) in patients (N=167) with PD-L1 expression ≥1%, 0.82 (95% CI: 0.44, 1.54) in patients (N=135) with PD-L1 expression <1%, and 0.50 (95% CI: 0.34, 0.73) in patients (N=488) with indeterminate/not reported/not evaluable PD-L1 expression.
Intravenous formulation
Randomised phase 3 study of nivolumab vs ipilimumab 10 mg/kg (CA209238)
The safety and efficacy of nivolumab 3 mg/kg as a single agent for the treatment of patients with completely resected melanoma were evaluated in a phase 3, randomised, double-blind study (CA209238). The study included adult patients, who had an ECOG performance status score of 0 or 1, with Stage IIIB/C or Stage IV American Joint Committee on Cancer (AJCC), 7th edition, histologically confirmed melanoma that is completely surgically resected. Per the AJCC 8th edition, this corresponds to patients with lymph node involvement or metastases. Patients were enrolled regardless of their tumour PD-L1 status. Patients with prior autoimmune disease, and any condition requiring systemic treatment with either corticosteroids (≥10 mg daily prednisone or equivalent) or other immunosuppressive medications, as well as patients with prior therapy for melanoma (except patients with surgery, adjuvant radiotherapy after neurosurgical resection for lesions of the central nervous system, and prior adjuvant interferon completed ≥6 months prior to randomisation) prior therapy with, anti-PD-1, anti-PD-L1, anti-PD-L2, anti-CD137, or anti CTLA-4 antibody (including ipilimumab or any other antibody or drug specifically targeting T cell co-stimulation or checkpoint pathways), were excluded from the study.
A total of 906 patients were randomised to receive either nivolumab 3 mg/kg (n=453) administered every 2 weeks or ipilimumab 10 mg/kg (n=453) administered every 3 weeks for 4 doses then every 12 weeks beginning at week 24 for up to 1 year. Randomisation was stratified by tumour PD-L1expression (≥5% vs. <5%/indeterminate), and stage of disease per the AJCC staging system. Tumour assessments were conducted every 12 weeks for the first 2 years then every 6 months thereafter. The primary endpoint was recurrence-free survival (RFS). RFS, assessed by investigator, was defined as the time between the date of randomisation and the date of first recurrence (local, regional, or distant metastasis), new primary melanoma, or death due to any cause, whichever occurred first.
Baseline characteristics were generally balanced between the two groups. The median age was 55 years (range: 18-86), 58% were men, and 95% were white. Baseline ECOG performance status score was 0 (90%) or 1 (10%). The majority of patients had AJCC Stage III disease (81%), and 19% had Stage IV disease. Forty-eight percent of patients had macroscopic lymph nodes and 32% had tumour ulceration. Forty-two percent of patients were BRAF V600 mutation positive while 45% were BRAF wild type and 13% BRAF were status unknown. For tumour PD-L1 expression, 34% of patients had PD-L1 expression ≥5% and 62% had <5% as determined by clinical trial assay. Among patients with quantifiable tumour PD-L1 expression, the distribution of patients was balanced across the treatment groups. Tumour PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay.
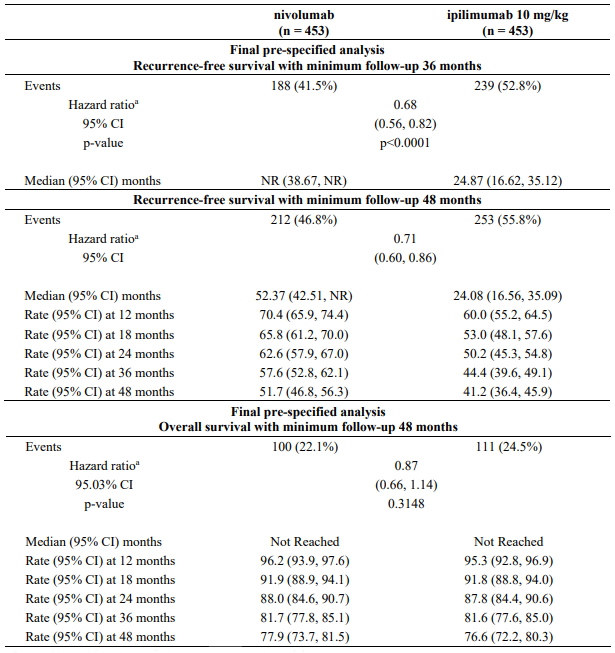
At a primary pre-specified interim analysis (minimum follow-up 18 months) a statistically significant improvement in RFS with nivolumab compared to ipilimumab with HR of 0.65 (97.56% CI: 0.51, 0.83; stratified log-rank p<0.0001) was demonstrated. At an updated descriptive RFS analysis, with minimum follow-up of 24 months RFS improvement was confirmed with HR of 0.66 (95% Cl: 0.54, 0.81; p<0.0001) and OS was not mature. Efficacy results with minimum follow-up of 36 months (RFS pre-specified final analysis) and 48 months (OS pre-specified final analysis) are shown in Table 15 and Figure 2 and 3 (all randomised population).
Table 15. Efficacy results (CA209238):
a Derived from a stratified proportional hazards model.
With a minimum follow-up of 36 months, the trial demonstrated a statistically significant improvement in RFS for patients randomised to the nivolumab arm compared with the ipilimumab 10 mg/kg arm. RFS benefit was consistently demonstrated across subgroups, including tumour PD-L1 expression, BRAF status, and stage of disease. With a minimum follow up of 48 months, shown in Figure 2, the trial continued to demonstrate improvement in RFS in the nivolumab arm compared with the ipilimumab arm. RFS benefit was sustained across all subgroups.
Figure 2. Recurrence-free survival (CA209238):
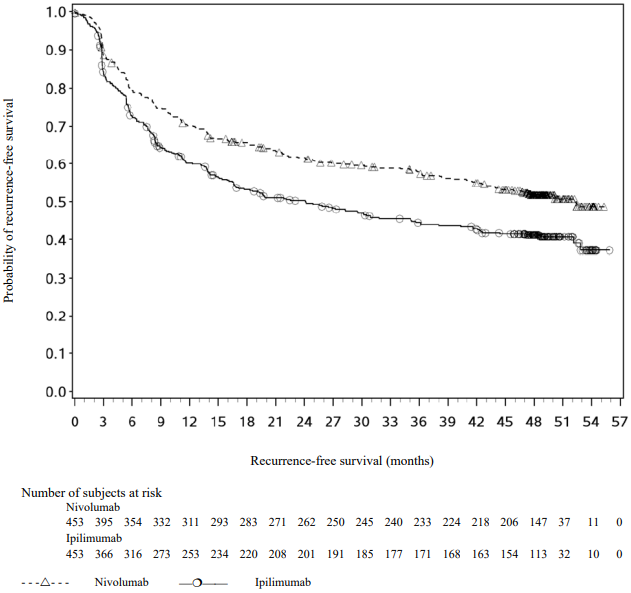
Figure 3. Overall survival (CA209238):
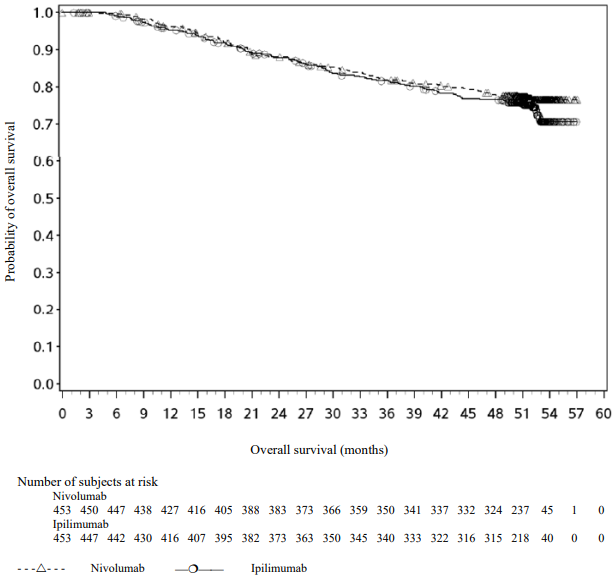
With a minimum follow-up of 48 months, shown in Figure 10, median OS was not reached in either group (HR = 0.87, 95.03% CI: 0.66, 1.14; p-value: 0.3148). The overall survival data are confounded by the effects of effective subsequent anti-cancer therapies. Subsequent systemic therapy was received by 33% and 42% of patients in the nivolumab and ipilimumab arms, respectively. Subsequent immunotherapy (including anti-PD1 therapy, anti-CTLA-4 antibody, or other immunotherapy) was received by 23% and 34% of patients in the nivolumab and ipilimumab arms, respectively.
Quality of life (QoL) with nivolumab remained stable and close to baseline values during treatment, as assessed by valid and reliable scales like the European Organisation for Research and Treatment of Cancer (EORTC) QLQ-C30 and the EQ-5D utility index and visual analog scale (VAS).
Treatment of advanced melanoma
Intravenous formulation
Randomised phase 3 study vs. dacarbazine (CA209066):
The safety and efficacy of nivolumab 3 mg/kg for the treatment of advanced (unresectable or metastatic) melanoma were evaluated in a phase 3, randomised, double-blind study (CA209066). The study included adult patients (18 years or older) with confirmed, treatment-naive, Stage III or IV BRAF wild-type melanoma and an ECOG performance-status score of 0 or 1. Patients with active autoimmune disease, ocular melanoma, or active brain or leptomeningeal metastases were excluded from the study.
A total of 418 patients were randomised to receive either nivolumab (n=210) administered intravenously over 60 minutes at 3 mg/kg every 2 weeks or dacarbazine (n=208) at 1000 mg/m² every 3 weeks. Randomisation was stratified by tumour PD-L1 status and M stage (M0/M1a/M1b versus M1c). Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. Treatment after disease progression was permitted for patients who had a clinical benefit and did not have substantial adverse events with the study drug, as determined by the investigator. Tumour assessments, according to the Response Evaluation Criteria in Solid Tumours (RECIST), version 1.1, were conducted 9 weeks after randomisation and continued every 6 weeks for the first year and then every 12 weeks thereafter. The primary efficacy outcome measure was OS. Key secondary efficacy outcome measures were investigator-assessed PFS and objective response rate (ORR).
Baseline characteristics were balanced between the two groups. The median age was 65 years (range: 18-87), 59% were men, and 99.5% were white. Most patients had ECOG performance score of 0 (64%) or 1 (34%). Sixty-one percent of patients had M1c stage disease at study entry. Seventy-four percent of patients had cutaneous melanoma, and 11% had mucosal melanoma; 35% of patients had PD-L1 positive melanoma (≥5% tumour cell membrane expression). Sixteen percent of patients had received prior adjuvant therapy; the most common adjuvant treatment was interferon (9%). Four percent of patients had a history of brain metastasis, and 37% of patients had a baseline LDH level greater than ULN at study entry.
The Kaplan-Meier curves for OS are shown in Figure 4.
Figure 4. Kaplan-Meier curves of OS (CA209066):
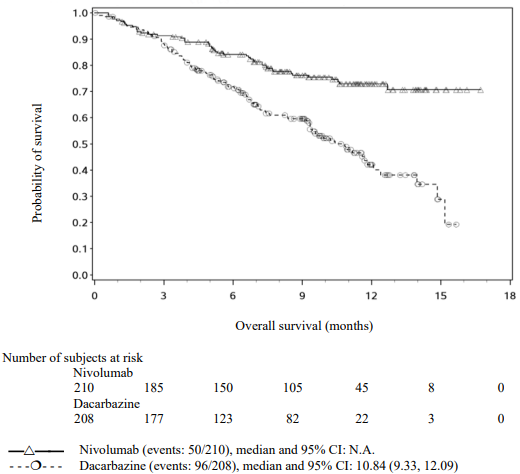
The observed OS benefit was consistently demonstrated across subgroups of patients including baseline ECOG performance status, M stage, history of brain metastases, and baseline LDH level. Survival benefit was observed regardless of whether patients had tumours that were designated PD-L1 negative or PD-L1 positive (tumour membrane expression cut off of 5% or 10%).
Data available indicate that the onset of nivolumab effect is delayed such that benefit of nivolumab above chemotherapy may take 2-3 months.
Efficacy results are shown in Table 16.
Table 16. Efficacy results (CA209066):

"+" denotes a censored observation.
Intravenous formulation
Randomised phase 3 study vs. chemotherapy (CA209037)
The safety and efficacy of nivolumab 3 mg/kg for the treatment of advanced (unresectable or metastatic) melanoma were evaluated in a phase 3, randomised, open-label study (CA209037). The study included adult patients who had progressed on or after ipilimumab and if BRAF V600 mutation positive had also progressed on or after BRAF kinase inhibitor therapy. Patients with active autoimmune disease, ocular melanoma, active brain or leptomeningeal metastases or a known history of prior ipilimumab-related high-grade (Grade 4 per CTCAE v4.0) adverse reactions, except for resolved nausea, fatigue, infusion reactions, or endocrinopathies, were excluded from the study.
A total of 405 patients were randomised to receive either nivolumab (n=272) administered intravenously over 60 minutes at 3 mg/kg every 2 weeks or chemotherapy (n=133) which consisted of the investigator's choice of either dacarbazine (1000 mg/m² every 3 weeks) or carboplatin (AUC 6 every 3 weeks) and paclitaxel (175 mg/m² every 3 weeks). Randomisation was stratified by BRAF and tumour PD-L1 status and best response to prior ipilimumab.
The co-primary efficacy outcome measures were confirmed ORR in the first 120 patients treated with nivolumab, as measured by independent radiology review committee (IRRC) using RECIST, version 1.1, and comparison of OS of nivolumab to chemotherapy. Additional outcome measures included duration and timing of response.
The median age was 60 years (range: 23-88). Sixty-four percent of patients were men and 98% were white. ECOG performance scores were 0 for 61% of patients and 1 for 39% of patients. The majority (75%) of patients had M1c stage disease at study entry. Seventy-three percent of patients had cutaneous melanoma and 10% had mucosal melanoma. The number of prior systemic regimen received was 1 for 27% of patients, 2 for 51% of patients, and >2 for 21% of patients. Twenty-two percent of patients had tumours that tested BRAF mutation positive and 50% of patients had tumours that were considered PD-L1 positive. Sixty-four percent of patients had no prior clinical benefit (CR/PR or SD) on ipilimumab. Baseline characteristics were balanced between groups except for the proportions of patients who had a history of brain metastasis (19% and 13% in the nivolumab group and chemotherapy group, respectively) and patients with LDH greater than ULN at baseline (51% and 35%, respectively).
At the time of this final ORR analysis, results from 120 nivolumab-treated patients and 47 chemotherapy-treated patients who had a minimum of 6 months of follow-up were analysed. Efficacy results are presented in Table 17.
Table 17. Best overall response, time and duration of response (CA209037):
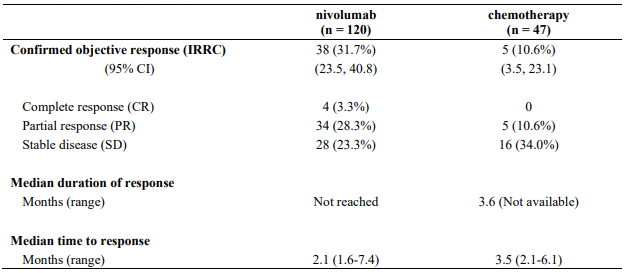
Data available indicate that the onset of nivolumab effect is delayed such that benefit of nivolumab above chemotherapy may take 2-3 months.
Updated analysis (24-month follow-up)
Among all randomised patients, the ORR was 27.2% (95% CI: 22.0, 32.9) in the nivolumab group and 9.8% (95% CI: 5.3, 16.1) in the chemotherapy group. Median durations of response were 31.9 months (range: 1.4+ - 31.9) and 12.8 months (range: 1.3+ - 13.6+), respectively. The PFS HR for nivolumab vs. chemotherapy was 1.03 (95% CI: 0.78, 1.36). The ORR and PFS were assessed by IRRC per RECIST version 1.1.
There was no statistically significant difference between nivolumab and chemotherapy in the final OS analysis. The primary OS analysis was not adjusted to account for subsequent therapies, with 54 (40.6%) patients in the chemotherapy arm subsequently receiving an anti-PD1 treatment. OS may be confounded by dropout, imbalance of subsequent therapies and differences in baseline factors. More patients in the nivolumab arm had poor prognostic factors (elevated LDH and brain metastases) than in the chemotherapy arm.
Efficacy by BRAF status: Objective responses to nivolumab (according to the definition of the co-primary endpoint) were observed in patients with or without BRAF mutation-positive melanoma. The ORRs in the BRAF mutation-positive subgroup were 17% (95% CI: 8.4, 29.0) for nivolumab and 11% (95% CI: 2.4, 29.2) for chemotherapy, and in the BRAF wild-type subgroup were 30% (95% CI: 24.0, 36.7) and 9% (95% CI: 4.6, 16.7), respectively.
The PFS HRs for nivolumab vs. chemotherapy were 1.58 (95% CI: 0.87, 2.87) for BRAF mutation-positive patients and 0.82 (95% CI: 0.60, 1.12) for BRAF wild-type patients. The OS HRs for nivolumab vs. chemotherapy were 1.32 (95% CI: 0.75, 2.32) for BRAF mutation-positive patients and 0.83 (95% CI: 0.62, 1.11) for BRAF wild-type patients.
Efficacy by tumour PD-L1 expression: Objective responses to nivolumab were observed regardless of tumour PD-L1 expression. However, the role of this biomarker (tumour PD-L1 expression) has not been fully elucidated.
In patients with tumour PD-L1 expression ≥1%, ORR was 33.5% for nivolumab (n=179; 95% CI: 26.7, 40.9) and 13.5% for chemotherapy (n=74; 95% CI: 6.7, 23.5). In patients with tumour PD-L1 expression <1%, ORR per IRRC was 13.0% (n=69; 95% CI: 6.1, 23.3) and 12.0% (n=25; 95% CI: 2.5, 31.2), respectively.
The PFS HRs for nivolumab vs. chemotherapy were 0.76 (95% CI: 0.54, 1.07) in patients with tumour PD-L1 expression ≥1% and 1.92 (95% CI: 1.05, 3.5) in patients with tumour PD-L1 expression <1%.
The OS HRs for nivolumab vs. chemotherapy were 0.69 (95% CI: 0.49, 0.96) in patients with tumour PD-L1 expression ≥1% and 1.52 (95% CI: 0.89, 2.57) in patients with tumour PD-L1 expression <1%.
These subgroup analyses should be interpreted with caution given the small size of the subgroups and lack of statistically significant difference in OS in the all randomised population.
Intravenous formulation
Open-label phase 1 dose-escalation study (MDX1106-03)
The safety and tolerability of nivolumab were investigated in a phase 1, open-label dose-escalation study in various tumour types, including malignant melanoma. Of the 306 previously treated patients enrolled in the study, 107 had melanoma and received nivolumab at a dose of 0.1 mg/kg, 0.3 mg/kg, 1 mg/kg, 3 mg/kg, or 10 mg/kg for a maximum of 2 years. In this patient population, objective response was reported in 33 patients (31%) with a median duration of response of 22.9 months (95% CI: 17.0, NR). The median PFS was 3.7 months (95% CI: 1.9, 9.3). The median OS was 17.3 months (95% CI: 12.5, 37.8), and the estimated OS rates were 42% (95% CI: 32, 51) at 3 years, 35% (95% CI: 26, 44) at 4 years, and 34% (95% CI: 25, 43) at 5 years (minimum follow-up of 45 months).
Intravenous formulation
Single-arm phase 2 study (CA209172)
Study CA209172 was a single-arm, open label study of nivolumab monotherapy in patients with stage III (unresectable) or stage IV metastatic melanoma after prior treatment containing an anti-CTLA-4 monoclonal antibody. Safety was the primary endpoint and efficacy was a secondary endpoint. Of the 1008 treated patients, 103 (10%) had ocular/uveal melanoma, 66 (7%) had an ECOG performance score of 2, 165 (16%) had asymptomatic treated and untreated CNS metastases, 13 (1.3%) had treated leptomeningeal metastases, 25 (2%) had autoimmune disease, and 84 (8%) had Grade 3-4 immune-related AEs with prior anti-CTLA-4 therapy. No new safety signals were identified in all treated patients and the overall safety profile of nivolumab was similar across subgroups. Efficacy results based on investigator-assessed response rates at week 12 are presented in Table 18 below.
Table 18. Response rate at week 12 - all response evaluable patients and by subgroup (CA209172):
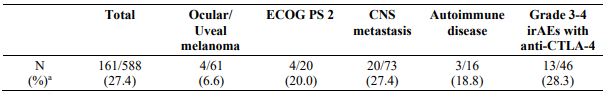
a Responses were assessed per RECIST 1.1 for 588/1008 (58.3%) of patients who continued treatment through week 12 and had a follow-up scan at week 12.
Intravenous formulation
Randomised phase 3 study of nivolumab in combination with ipilimumab or nivolumab as monotherapy vs. ipilimumab as monotherapy (CA209067)
The safety and efficacy of nivolumab 1 mg/kg in combination with ipilimumab 3 mg/kg or nivolumab 3 mg/kg vs. ipilimumab 3 mg/kg monotherapy for the treatment of advanced (unresectable or metastatic) melanoma were evaluated in a phase 3, randomised, double-blind study (CA209067). The differences between the two nivolumab-containing groups were evaluated descriptively. The study included adult patients with confirmed unresectable Stage III or Stage IV melanoma. Patients were to have ECOG performance status score of 0 or 1. Patients who had not received prior systemic anticancer therapy for unresectable or metastatic melanoma were enrolled. Prior adjuvant or neoadjuvant therapy was allowed if it was completed at least 6 weeks prior to randomisation. Patients with active autoimmune disease, ocular/uveal melanoma, or active brain or leptomeningeal metastases were excluded from the study.
A total of 945 patients were randomised to receive nivolumab in combination with ipilimumab (n=314), nivolumab monotherapy (n=316), or ipilimumab monotherapy (n=315). Patients in the combination arm received nivolumab 1 mg/kg over 60 minutes and ipilimumab 3 mg/kg over 90 minutes administered intravenously every 3 weeks for the first 4 doses, followed by nivolumab 3 mg/kg as monotherapy every 2 weeks. Patients in the nivolumab monotherapy arm received nivolumab 3 mg/kg every 2 weeks. Patients in the comparator arm received ipilimumab 3 mg/kg and nivolumab-matched placebo intravenously every 3 weeks for 4 doses followed by placebo every 2 weeks. Randomisation was stratified by PD-L1 expression (≥5% vs. <5% tumour cell membrane expression), BRAF status, and M stage per the American Joint Committee on Cancer (AJCC) staging system. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. Tumour assessments were conducted 12 weeks after randomisation then every 6 weeks for the first year, and every 12 weeks thereafter. The primary outcome measures were progression-free survival and OS. ORR and the duration of response were also assessed.
Baseline characteristics were balanced across the three treatment groups. The median age was 61 years (range: 18 to 90 years), 65% of patients were men, and 97% were white. ECOG performance status score was 0 (73%) or 1 (27%). The majority of the patients had AJCC Stage IV disease (93%); 58% had M1c disease at study entry. Twenty-two percent of patients had received prior adjuvant therapy. Thirty-two percent of patients had BRAF mutation-positive melanoma; 26.5% of patients had PD-L1 ≥5% tumour cell membrane expression. Four percent of patients had a history of brain metastasis, and 36% of patients had a baseline LDH level greater than ULN at study entry. Among patients with quantifiable tumour PD-L1 expression, the distribution of patients was balanced across the three treatment groups. Tumour PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay.
At primary analysis (minimum follow-up 9 months) the median PFS was 6.9 months in the nivolumab group as compared with 2.9 months in the ipilimumab group (HR = 0.57, 99.5% CI: 0.43, 0.76; p<0.0001). The median PFS was 11.5 months in the nivolumab in combination with ipilimumab group, as compared with 2.9 months in the ipilimumab group (HR = 0.42, 99.5% CI: 0.31, 0.57; p<0.0001).
PFS results from descriptive analysis (with minimum follow up of 90 months) are shown in Figure 5 (all randomised population), Figure 6 (at the tumour PD-L1 5% cut off), and Figure 7 (at the tumour PD-L1 1% cut off).
Figure 5. Progression-free survival (CA209067):
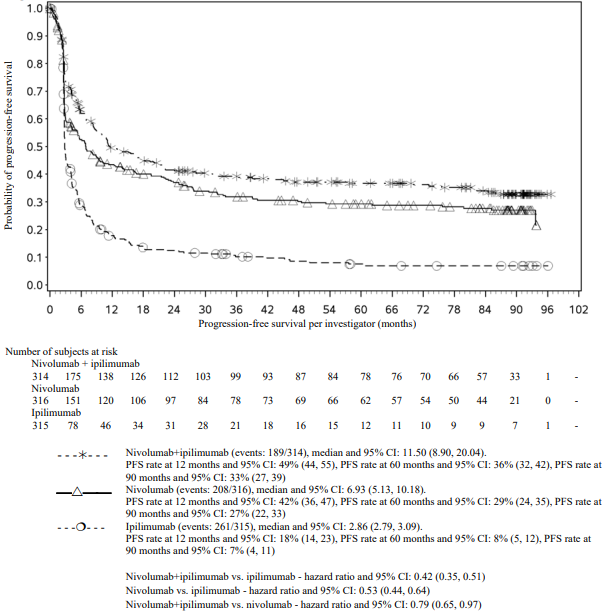
Figure 6. Progression-free survival by PD-L1 expression: 5% cut off (CA209067):
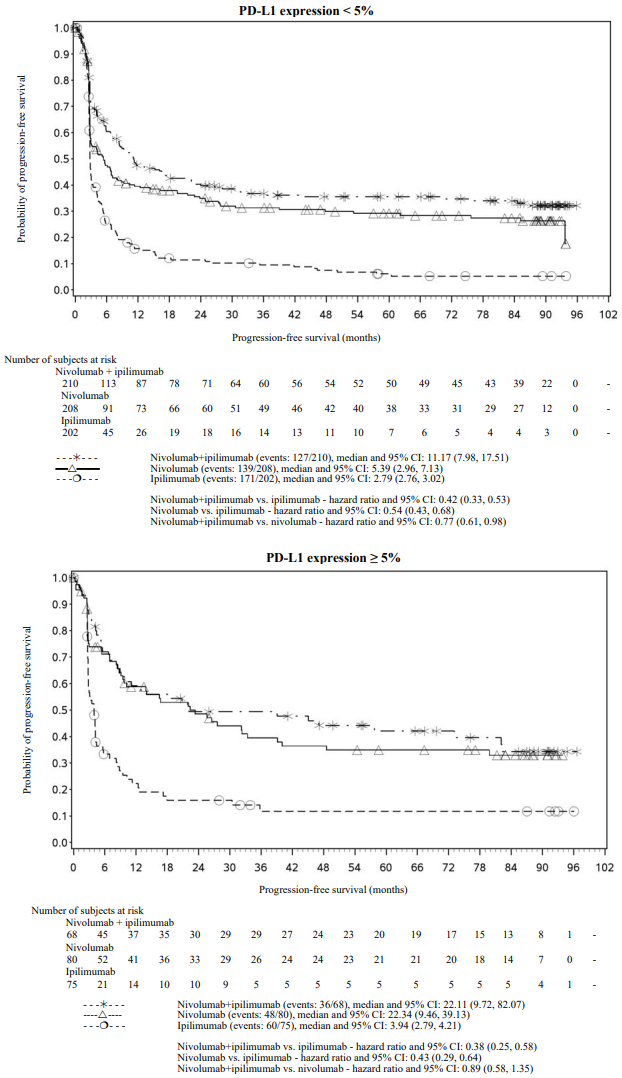
Figure 7. Progression-free survival by PD-L1 expression: 1% cut off (CA209067):
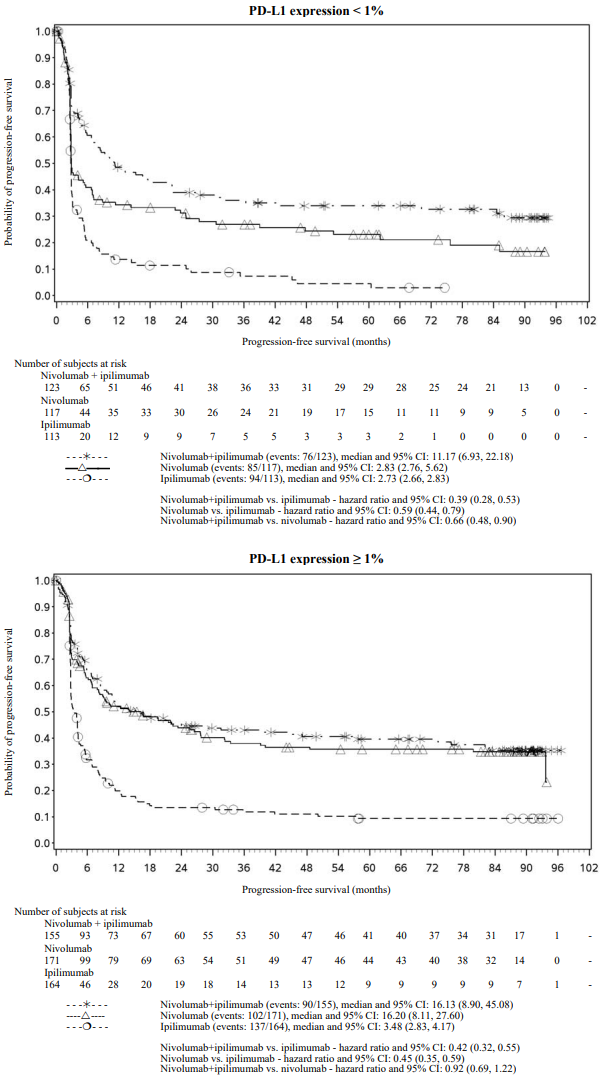
The final (primary) OS analysis occurred when all patients had a minimum follow-up of 28 months. At 28 months, median OS was not reached in the nivolumab group as compared with 19.98 months in the ipilimumab group (HR = 0.63, 98% CI: 0.48, 0.81; p-value: <0.0001). Median OS was not reached in the nivolumab in combination with ipilimumab group as compared with the ipilimumab group (HR = 0.55, 98% CI: 0.42, 0.72; p-value: <0.0001).
OS results at an additional descriptive analysis undertaken at a minimum follow-up of 90 months show outcomes consistent with the original primary analysis. OS results from this follow-up analysis are shown in Figure 8 (all randomised), Figure 9 and 10 (at the tumour PD-L1 5% and 1% cut off).
The OS analysis was not adjusted to account for subsequent therapies received. Subsequent systemic therapy was received by 36.0%, 49.1%, and 66.3% of patients in the combination, nivolumab monotherapy, and ipilimumab arms, respectively. Subsequent immunotherapy (including anti-PD1 therapy, anti-CTLA-4 antibody, or other immunotherapy) was received by 19.1%, 34.2%, and 48.3% of patients in the combination, nivolumab monotherapy, and ipilimumab arms, respectively.
Figure 8. Overall survival (CA209067) - Minimum follow-up of 90 months:
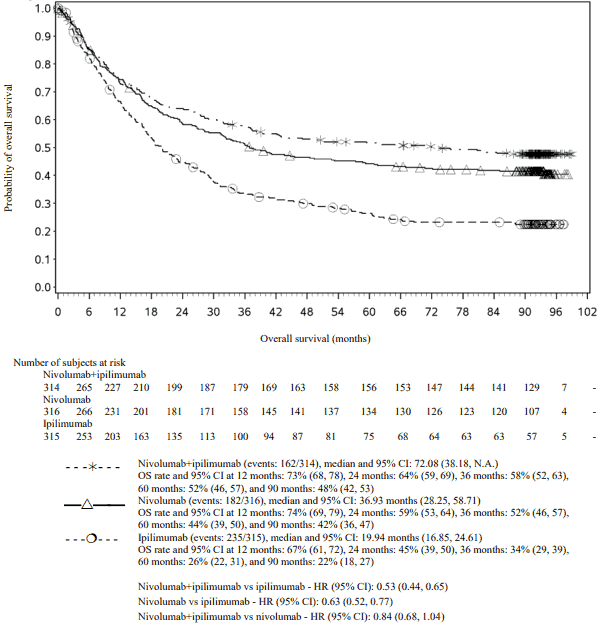
Figure 9. Overall survival by PD-L1 expression: 5% cut off (CA209067) - Minimum follow-up of 90 months:
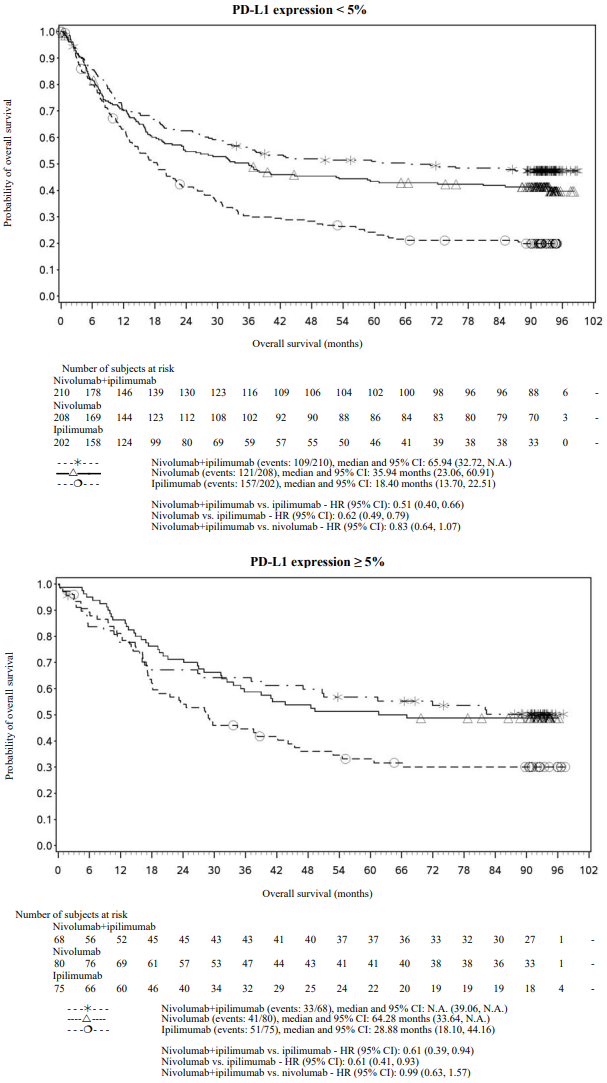
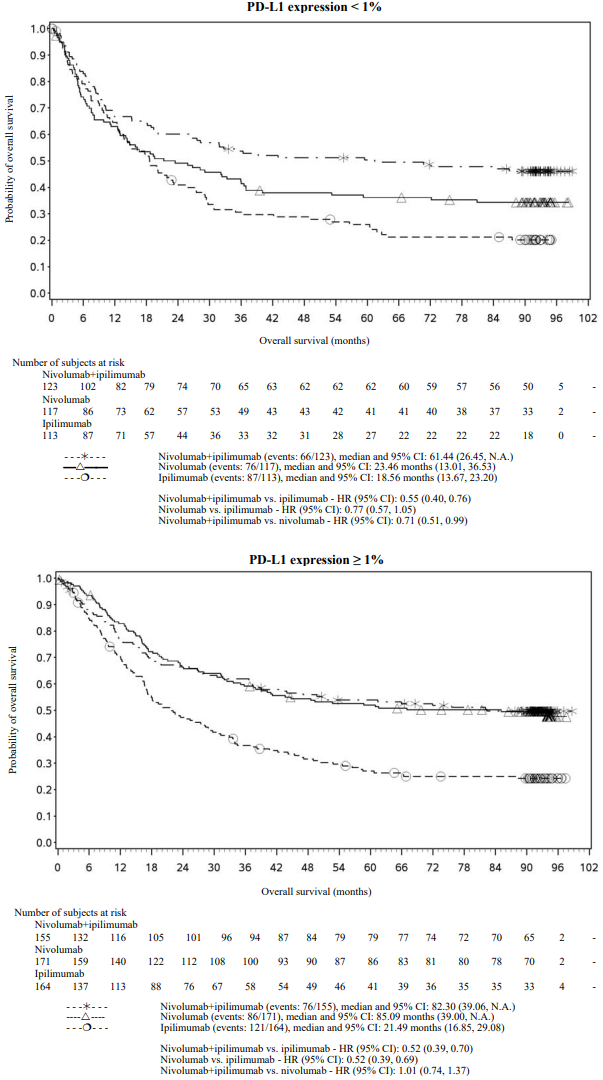
Figure 10. Overall survival by PD-L1 expression: 1% cut off (CA209067) - Minimum follow-up of 90 months:
Minimum follow-up for the analysis of ORR was 90 months. Responses are summarised in Table 19.
Table 19. Objective response (CA209067):
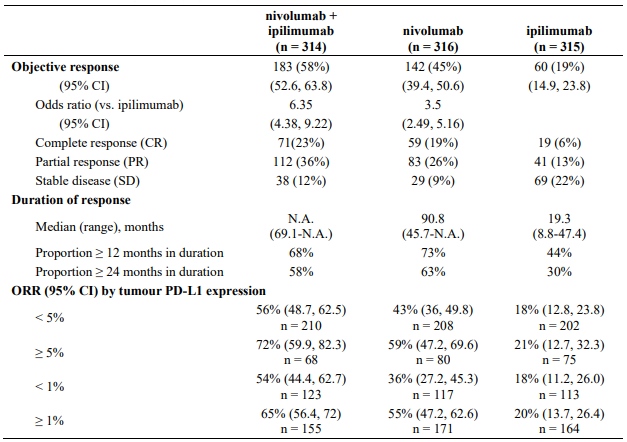
Both nivolumab-containing arms demonstrated a significant PFS and OS benefit and greater ORR compared with ipilimumab alone. The observed PFS results at 18 months of follow-up and ORR and OS results at 28 months of follow-up were consistently demonstrated across subgroups of patients including baseline ECOG performance status, BRAF status, M stage, age, history of brain metastases, and baseline LDH level. This observation was maintained with the OS results with a minimum follow-up of 90 months.
Among 131 patients who discontinued the combination due to adverse reaction after 28 months of follow-up, the ORR was 71% (93/131) with 20% (26/131) achieving a complete response and median OS was not reached.
Both nivolumab-containing arms demonstrated greater objective response rates than ipilimumab regardless of PD-L1 expression levels. ORRs were higher for the combination of nivolumab and ipilimumab relative to nivolumab monotherapy across tumour PD-L1 expression levels (Table 19) after 90 months of follow-up, with a best overall response of complete response correlating to an improved survival rate.
After 90 months of follow-up, median durations of response for patients with tumour PD-L1 expression level ≥5% were 78.19 months (range: 18.07-N.A.) in the combination arm,77.21 months (range: 26.25-N.A.) in the nivolumab monotherapy arm and 31.28 months (range: 6.08-N.A.) in the ipilimumab arm. At tumour PD-L1 expression <5%, median durations of response were not reached (range: 61.93-N.A.) in the combination arm, were 90.84 months (range: 50.43-N.A.) in the nivolumab monotherapy arm and 19.25 months (range: 5.32-47.44) in the ipilimumab monotherapy arm.
No clear cut off for PD-L1 expression can reliably be established when considering the relevant endpoints of tumour response and PFS and OS. Results from exploratory multivariate analyses identified patient and tumour characteristics (ECOG performance status, M stage, baseline LDH, BRAF mutation status, PD-L1 status, and gender) which might contribute to the survival outcome.
Efficacy by BRAF status:
After 90 months of follow-up, BRAF[V600] mutation-positive and BRAF wild-type patients randomised to nivolumab in combination with ipilimumab had a median PFS of 16.76 months (95% CI: 8.28, 32.0) and 11.7 months (95% CI: 7.0, 19.32), while those in the nivolumab monotherapy arm had a median PFS of 5.62 months (95% CI: 2.79, 9.46) and 8.18 months (95% CI: 5.13, 19.55), respectively. BRAF[V600] mutation-positive and BRAF wild-type patients randomised to ipilimumab monotherapy had a median PFS of 3.09 months (95% CI: 2.79, 5.19) and 2.83 months (95% CI: 2.76, 3.06), respectively.
After 90 months of follow-up, BRAF[V600] mutation-positive and BRAF wild-type patients randomised to nivolumab in combination with ipilimumab had an ORR of 67.0% (95% CI: 57.0, 75.9; n=103) and 54.0% (95% CI: 47.1, 60.9; n=211), while those in the nivolumab monotherapy arm had an ORR of 37.87% (95% CI: 28.2, 48.1; n=98) and 48.2% (95% CI: 41.4, 55.0; n=218), respectively. BRAF[V600] mutation-positive and BRAF wild-type patients randomised to ipilimumab monotherapy had an ORR of 23.0% (95% CI: 15.2, 32.5; n=100) and 17.2% (95% CI: 12.4, 22.9; n=215).
After 90 months of follow-up, in BRAF [V600] mutation-positive patients median OS was not reached in the combination arm and 45.5 months in the nivolumab monotherapy arm. Median OS for BRAF [V600] mutation-positive patients in the ipilimumab monotherapy arm was 24.6 months. In BRAF wild-type patients median OS was 39.06 months in the combination arm, 34.37 months in the nivolumab monotherapy arm and 18.5 months in the ipilimumab monotherapy arm. The OS HRs for nivolumab in combination with ipilimumab vs. nivolumab monotherapy were 0.66 (95% CI: 0.44, 0.98) for BRAF[V600] mutation-positive patients and 0.95 (95% CI: 0.74, 1.22) for BRAF wild-type patients.
Intravenous formulation
Randomised phase 2 study of nivolumab in combination with ipilimumab and ipilimumab (CA209069)
Study CA209069 was a randomised, Phase 2, double-blind study comparing the combination of nivolumab and ipilimumab with ipilimumab alone in 142 patients with advanced (unresectable or metastatic) melanoma with similar inclusion criteria to study CA209067 and the primary analysis in patients with BRAF wild-type melanoma (77% of patients). Investigator assessed ORR was 61% (95% CI: 48.9, 72.4) in the combination arm (n=72) versus 11% (95% CI: 3.0, 25.4) for the ipilimumab arm (n=37). The estimated 2 and 3 year OS rates were 68% (95% CI: 56, 78) and 61% (95% CI: 49, 71), respectively, for the combination (n=73) and 53% (95% CI: 36, 68) and 44% (95% CI: 28, 60), respectively, for ipilimumab (n=37).
Non-small cell lung cancer
Neoadjuvant and adjuvant treatment of NSCLC
Intravenous formulation
Randomised, double-blind, phase 3 study of neoadjuvant nivolumab in combination with platinum-based chemotherapy vs. platinum-based chemotherapy and adjuvant nivolumab monotherapy vs. placebo (CA20977T):
The safety and efficacy of nivolumab in combination with platinum-based chemotherapy for 4 cycles, followed by nivolumab monotherapy, were evaluated in a randomised, double-blind study (CA20977T). The study included patients with ECOG performance status 0 or 1 whose tumours were resectable, suspected or histologically confirmed Stage IIA (>4 cm) to IIIB (T3-T4 N2) NSCLC (per the 8th edition American Joint Committee on Cancer (AJCC) Staging Manual). Patients were enrolled regardless of their tumour PD-L1 status.
The following selection criteria define patients with high risk of recurrence who are included in the therapeutic indication and are reflective of a patient population with stage IIA-IIIB disease according to the 8th edition AJCC/UICC staging criteria: any patient with a tumour size >4 cm; any patient with N1 or N2 disease (regardless of primary tumour size); patients with multiple tumour nodules in either the same lobe or different ipsilateral lobes; patients with tumours that are invasive of thoracic structures (directly invade visceral pleura, parietal pleura, chest wall, diaphragm, phrenic nerve, mediastinal pleura, parietal pericardium, mediastinum, heart, great vessels, trachea, recurrent laryngeal nerve, oesophagus, vertebral body, carina); or tumours that involve the main bronchus; or tumours that are associated with atelectasis or obstructive pneumonitis that extends to the hilar region or involves the entire lung.
Patients with unresectable or metastatic NSCLC, EGFR mutations or known ALK translocations (testing for ALK translocations was not mandatory at study entry), brain metastasis, Grade 2 or greater peripheral neuropathy, interstitial lung disease or active, non-infectious pneumonitis (symptomatic and/or requiring treatment), active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study.
A total of 461 patients were randomised to receive either nivolumab in combination with platinum-based chemotherapy followed by nivolumab monotherapy (n=229) or platinum-based chemotherapy followed by placebo (n=232). In the neoadjuvant phase, patients received either nivolumab 360 mg administered intravenously over 30 minutes and platinum-doublet chemotherapy administered every 3 weeks, or placebo and platinum-doublet chemotherapy administered every 3 weeks until disease progression or unacceptable toxicity, for up to 4 cycles. Patients in both treatment arms could receive post-operative radiation therapy (PORT) as standard of care. In the adjuvant phase, within 90 days after the surgery patients received either nivolumab 480 mg administered intravenously over 30 minutes every 4 weeks, or placebo administered every 4 weeks for until disease progression, recurrence, or unacceptable toxicity, for up to 13 cycles. Platinum-based chemotherapy consisted of paclitaxel 175 mg/m² or 200 mg/m² and carboplatin AUC 5 or AUC 6 (any histology); pemetrexed 500 mg/m², and cisplatin 75 mg/m² or carboplatin AUC 5 or AUC 6 (non-squamous histology); or cisplatin 75 mg/m² and docetaxel 75 mg/m² (squamous histology).
Stratification factors for randomisation were tumour PD-L1 expression level (≥1% versus <1% versus indeterminate/not evaluable), disease stage (Stage II versus Stage III), and tumour histology (squamous versus nonsquamous). Tumour PD-L1 expression levels were assessed using the PD-L1 IHC 28-8 pharmDx test. Tumour assessments were performed at baseline, within 14 days after the last dose of neoadjuvant treatment and before surgery, within 7 days prior to the start of adjuvant treatment after surgery, every 12 weeks after the first dose of adjuvant treatment for 2 years, then every 24 weeks for up to 5 years until disease recurrence or progression was confirmed by BICR.
Among the 442 patients in CA20977T, 256 (58%) had tumour PD-L1 expression ≥1% based on the PD-L1 IHC 28-8 pharmDx test. The median age was 66 years (range: 35 to 86) with 55% of patients ≥65 years and 7% of patients ≥75 years, 69% were White, 28% were Asian, 2% were Black, and 75% were male. Baseline ECOG performance status was 0 (59%) or 1 (41%); 36% had stage II and 63% had stage III disease; 24% were N1 and 39% were N2; 25% were single-station and 14% were multistation; 61% had tumours with squamous histology and 39% had tumours with non-squamous histology; and 91% were former/current smokers.
Seventy-eight percent of patients in the neoadjuvant nivolumab in combination with platinum-doublet chemotherapy followed by adjuvant nivolumab arm had definitive surgery compared to 77% of patients in the neoadjuvant placebo and platinum-doublet chemotherapy followed by placebo arm. Approximately 5% of patients in each treatment arm received PORT.
The primary efficacy outcome measure was event-free survival (EFS) based on BICR assessment. Additional efficacy outcome measures included overall survival (OS), pathologic complete response (pCR) and major pathologic response as evaluated by blinded independent pathology review (BIPR).
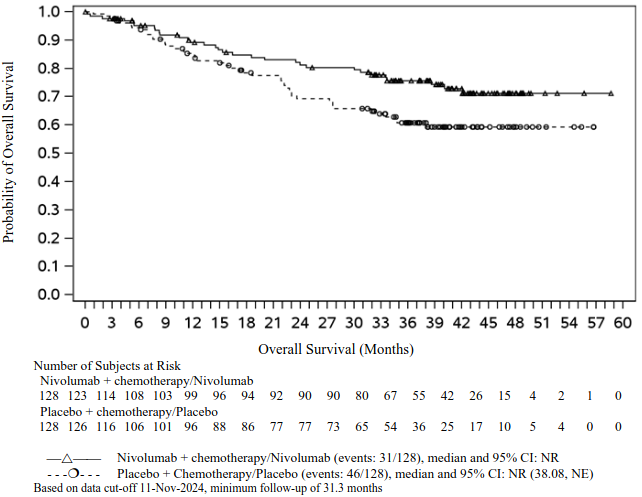
In a pre-specified interim analysis in all randomised patients with a median follow-up of 25.4 months (range: 15.7-44.2 months), the study demonstrated statistically significant improvement of EFS. Median EFS was not reached (95% CI: 28.94, NE) in the nivolumab in combination with chemotherapy/nivolumab arm and 18.43 months (95% CI: 13.63, 28.06) in the placebo with chemotherapy/placebo arm (HR = 0.58, 97.36% CI: 0.42, 0.81; stratified log-rank p-value 0.00025). In a pre-specified interim analysis in all randomised patients with a median follow-up of 41 months (range: 31.3-59.8 months), median OS was not reached in both the nivolumab in combination with chemotherapy/nivolumab arm and in the placebo with chemotherapy/placebo arm (HR = 0.85, 97.63% CI: 0.58, 1.25).
Exploratory subgroup analysis by tumour PD-L1 expression:
EFS for the subgroup of patients with tumour PD-L1 expression ≥1%, with a median follow-up of 41 months (range: 31.3-59.8 months), are presented in Table 20 and Figure 11.
Table 20. Efficacy results in patients with tumour PD-L1 ≥1% (CA20977T):
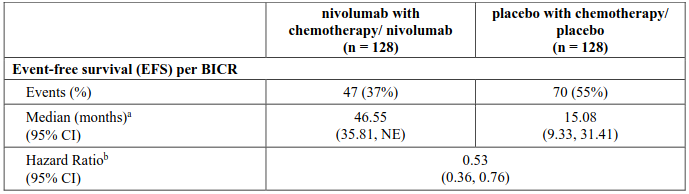
NE = non-estimable
Minimum follow-up for EFS was 31.3 months; data cut-off: 11-Nov-2024.
a Kaplan-Meier estimate.
b Based on an unstratified Cox proportional hazard model.
Figure 11. Kaplan-Meier curves of EFS in patients with tumour PD-L1 ≥1% (CA20977T):
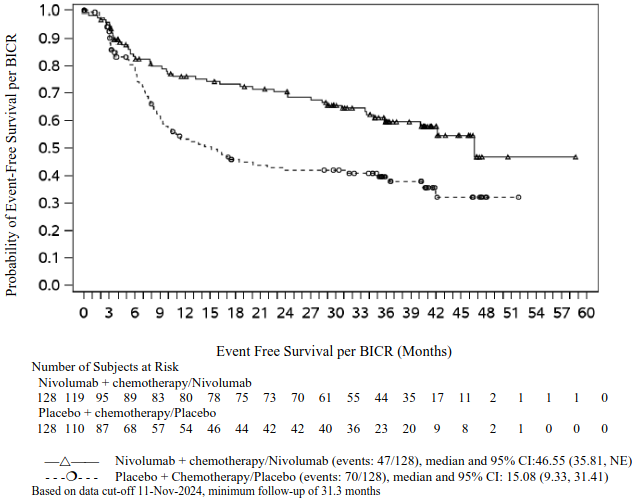
At the time of the updated EFS analysis, an interim analysis for OS was performed (minimum follow-up of 31.3 months). The exploratory, descriptive HR for OS in patients with tumour PD-L1 expression ≥1% was 0.61 (95% CI: 0.39, 0.97) for the nivolumab in combination with chemotherapy/nivolumab arm vs. the placebo with chemotherapy/placebo arm. The Kaplan-Meier curves for OS for the subgroup of patients with tumour PD-L1 expression ≥1% are shown in Figure 12.
Figure 12. Kaplan-Meier curves of OS in patients with tumour PD-L1 ≥1% (CA20977T):
Treatment of NSCLC after prior chemotherapy
Squamous NSCLC
Intravenous formulation
Randomised phase 3 study vs. docetaxel (CA209017):
The safety and efficacy of nivolumab 3 mg/kg as a single agent for the treatment of advanced or metastatic squamous NSCLC were evaluated in a phase 3, randomised, open-label study (CA209017). The study included patients (18 years or older) who have experienced disease progression during or after one prior platinum doublet-based chemotherapy regimen and an ECOG performance status score of 0 or 1. Patients were enrolled regardless of their tumour PD-L1 status. Patients with active autoimmune disease, symptomatic interstitial lung disease, or active brain metastases were excluded from the study. Patients with treated brain metastases were eligible if neurologically returned to baseline at least 2 weeks prior to enrolment, and either off corticosteroids, or on a stable or decreasing dose of <10 mg daily prednisone equivalents.
A total of 272 patients were randomised to receive either nivolumab 3 mg/kg (n=135) administered intravenously over 60 minutes every 2 weeks or docetaxel (n=137) 75 mg/m² every 3 weeks. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. Tumour assessments, according to the RECIST, version 1.1, were conducted 9 weeks after randomisation and continued every 6 weeks thereafter. The primary efficacy outcome measure was OS. Key secondary efficacy outcome measures were investigator-assessed ORR and PFS. In addition, symptom improvement and overall health status were assessed using the Lung cancer symptom score (LCSS) average symptom burden index and the EQ-5D Visual Analogue Scale (EQ-VAS), respectively.
Baseline characteristics were generally balanced between the two groups. The median age was 63 years (range: 39-85) with 44% ≥65 years of age and 11% ≥75 years of age. The majority of patients were white (93%) and male (76%). Thirty-one percent had progressive disease reported as the best response to their most recent prior regimen and 45% received nivolumab within 3 months of completing their most recent prior regimen. Baseline ECOG performance status score was 0 (24%) or 1 (76%).
The Kaplan-Meier curves for OS are shown in Figure 13.
Figure 13. Kaplan-Meier curves of OS (CA209017):
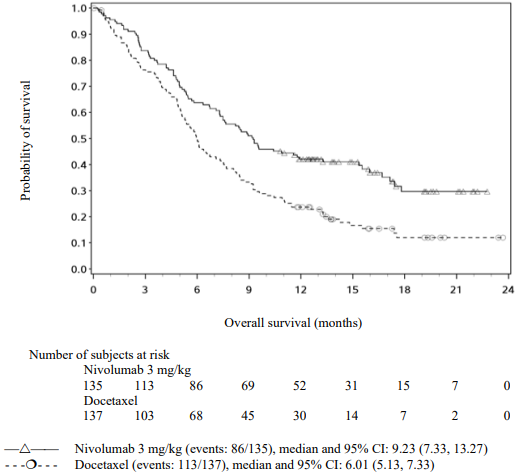
The observed OS benefit was consistently demonstrated across subgroups of patients. Survival benefit was observed regardless of whether patients had tumours that were designated PD-L1 negative or PD-L1 positive (tumour membrane expression cut off of 1%, 5% or 10%). However, the role of this biomarker (tumour PD-L1 expression) has not been fully elucidated. With a minimum of 62.6 months follow-up, OS benefit remains consistently demonstrated across subgroups.
Study CA209017 included a limited number of patients ≥75 years (11 in the nivolumab group and 18 in the docetaxel group). Nivolumab showed numerically less effect on OS (HR 1.85; 95% CI: 0.76, 4.51), PFS (HR = 1.76; 95%-CI: 0.77, 4.05) and ORR (9.1% vs. 16.7%). Because of the small sample size, no definitive conclusions can be drawn from these data.
Efficacy results are shown in Table 21.
Table 21. Efficacy results (CA209017):
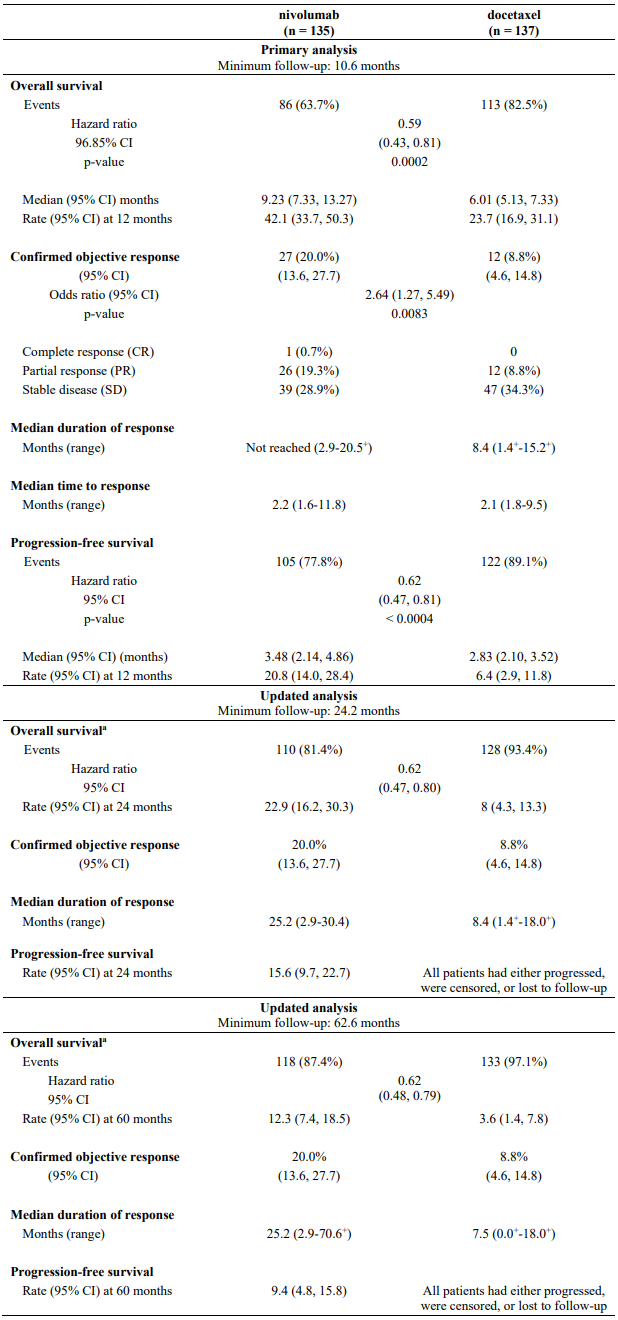
a Six patients (4%) randomised to docetaxel crossed over at any time to receive nivolumab treatment.
"+" Denotes a censored observation.
The rate of disease-related symptom improvement, as measured by LCSS, was similar between the nivolumab group (18.5%) and the docetaxel group (21.2%). The average EQ-VAS increased over time for both treatment groups, indicating better overall health status for patients remaining on treatment.
Intravenous formulation
Single-arm phase 2 study (CA209063):
Study CA209063 was a single-arm, open-label study conducted in 117 patients with locally advanced or metastatic squamous NSCLC after two or more lines of therapy; otherwise similar inclusion criteria as study CA209017 were applied. Nivolumab 3 mg/kg showed an ORR of 14.5% (95% CI: 8.7,22.2%), a median OS of 8.21 months (95% CI: 6.05,10.9), and a median PFS of 1.87 months (95% CI 1.77,3.15). The PFS was measured by RECIST, version 1.1. The estimated 1-year survival rate was 41%.
Intravenous formulation
Single-arm phase 2 study (CA209171):
Study CA209171 was a single-arm, open label study of nivolumab monotherapy in patients with previously treated advanced or metastatic squamous NSCLC. Safety was the primary endpoint and efficacy was a secondary endpoint. Of the 811 treated patients, 103 (13%) had an ECOG performance score of 2, 686 (85%) were <75 years old and 125 (15%) were ≥75 years old. No new safety signals were identified in all treated patients and the overall safety profile of nivolumab was similar across subgroups. Efficacy results based on investigator-assessed ORR are presented in Table 22 below.
Table 22. ORR based on response evaluable patients – total and by subgroup (CA209171):
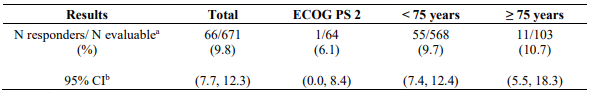
a includes confirmed and unconfirmed responses, scans were mandatory only at week 8/9 and week 52.
b CR+PR, confidence interval based on the Clopper and Pearson method
Non-squamous NSCLC
Intravenous formulation
Randomised phase 3 study vs. docetaxel (CA209057):
The safety and efficacy of nivolumab 3 mg/kg as a single agent for the treatment of advanced or metastatic non-squamous NSCLC were evaluated in a phase 3, randomised, open-label study (CA209057). The study included patients (18 years or older) who have experienced disease progression during or after one prior platinum doublet-based chemotherapy regimen which may have included maintenance therapy and who had an ECOG performance status score of 0 or 1. An additional line of TKI therapy was allowed for patients with known EGFR mutation or ALK translocation. Patients were enrolled regardless of their tumour PD-L1 status. Patients with active autoimmune disease, symptomatic interstitial lung disease, or active brain metastases were excluded from the study. Patients with treated brain metastases were eligible if neurologically returned to baseline at least 2 weeks prior to enrolment, and either off corticosteroids, or on a stable or decreasing dose of <10 mg daily prednisone equivalents.
A total of 582 patients were randomised to receive either nivolumab 3 mg/kg administered intravenously over 60 minutes every 2 weeks (n=292) or docetaxel 75 mg/m² every 3 weeks (n=290). Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. Tumour assessments were conducted according to the RECIST version 1.1. The primary efficacy outcome measure was OS. Key secondary efficacy outcome measures were investigator-assessed ORR and PFS. Additional prespecified subgroup analyses were conducted to evaluate the efficacy of tumour PD-L1 expression at predefined levels of 1%, 5% and 10%. Assessment according to discrete PD-L1 expression intervals were not included in the prespecified analyses due to the small sample sizes within the intervals.
Pre-study tumour tissue specimens were systematically collected prior to randomisation in order to conduct pre-planned analyses of efficacy according to tumour PD-L1 expression. Tumour PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay.
The median age was 62 years (range: 21 to 85) with 34% ≥65 years of age and 7% ≥75 years of age. The majority of patients were white (92%) and male (55%). Baseline ECOG performance status was 0 (31%) or 1 (69%). Seventy-nine percent of patients were former/current smokers.
The Kaplan-Meier curves for OS are shown in Figure 14.
Figure 14. Kaplan-Meier curves of OS (CA209057):
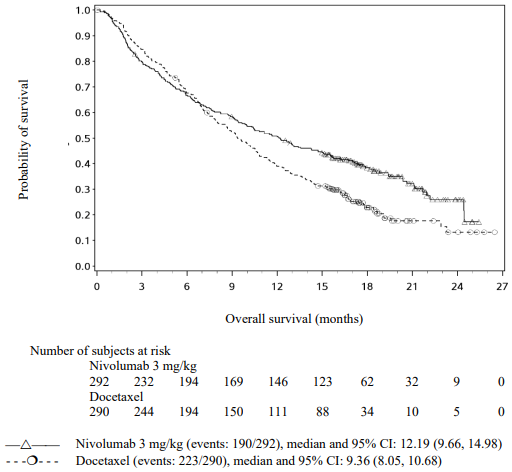
The trial demonstrated a statistically significant improvement in OS for patients randomised to nivolumab as compared with docetaxel at the prespecified interim analysis when 413 events were observed (93% of the planned number of events for final analysis). Efficacy results are shown in Table 23.
Table 23. Efficacy results (CA209057):
a Derived from a stratified proportional hazards model.
b P-value is derived from a log-rank test stratified by prior maintenance therapy and line of therapy; the corresponding O'Brien-Fleming efficacy boundary significance level is 0.0408.
c Sixteen patients (6%) randomised to docetaxel crossed over at any time to receive nivolumab treatment.
d Seventeen patients (6%) randomised to docetaxel crossed over at any time to receive nivolumab treatment.
"+" Denotes a censored observation.
Quantifiable tumour PD-L1 expression was measured in 79% of patients in the nivolumab group and 77% of patients in the docetaxel group. Tumour PD-L1 expression levels were balanced between the two treatment groups (nivolumab vs. docetaxel) at each of the predefined tumour PD-L1 expression levels of ≥1% (53% vs. 55%), ≥5% (41% vs. 38%), or ≥10% (37% vs. 35%).
Patients with tumour PD-L1 expression by all predefined expression levels in the nivolumab group demonstrated greater likelihood of improved survival compared to docetaxel, whereas survival was similar to docetaxel in patients with low or no tumour PD-L1 expression. In terms of ORR, increasing PD-L1 expression was associated with larger ORR. Comparable to the overall population, median duration of response was increased with nivolumab vs. docetaxel for patients with no PD-L1 expression (18.3 months vs. 5.6 months) and for patients with PD-L1 expression (16.0 months vs. 5.6 months).
Table 24 summarises results of ORR and OS by tumour PD-L1 expression.
Table 24. ORR and OS by tumour PD-L1 expression (CA209057):
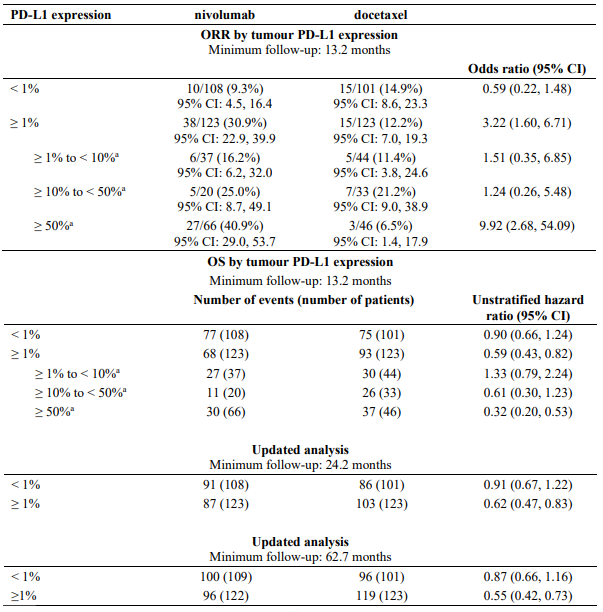
a Post-hoc analysis; results should be interpreted with caution as the subgroup samples sizes are small and, at the time of the analysis, the PD-L1 IHC 28-8 pharmDx assay was not analytically validated at the 10% or 50% expression levels.
A higher proportion of patients experienced death within the first 3 months in the nivolumab arm (59/292, 20.2%) as compared to the docetaxel arm (44/290, 15.2%). Results of a post-hoc, exploratory multivariate analysis indicated that nivolumab-treated patients with poorer prognostic features and/or aggressive disease when combined with lower (e.g., <50%) or no tumour PD-L1 expression may be at higher risk of death within the first 3 months.
In subgroup analyses, survival benefit compared to docetaxel was not shown for patients who were never-smokers or whose tumours harboured EGFR activating mutations; however, due to the small numbers of patients, no definitive conclusions can be drawn from these data.
Renal cell carcinoma (RCC)
Intravenous formulation
Randomised phase 3 study of nivolumab in combination with ipilimumab vs. sunitinib (CA209214)
The safety and efficacy of nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg for the treatment of advanced/metastatic RCC was evaluated in a phase 3, randomised, open-label study (CA209214). The study included patients (18 years or older) with previously untreated, advanced or metastatic renal cell carcinoma with a clear-cell component. The primary efficacy population included those intermediate/poor risk patients with at least 1 or more of 6 prognostic risk factors as per the International Metastatic RCC Database Consortium (IMDC) criteria (less than one year from time of initial renal cell carcinoma diagnosis to randomisation, Karnofsky performance status <80%, haemoglobin less than the lower limit of normal, corrected calcium of greater than 10 mg/dL, platelet count greater than the upper limit of normal, and absolute neutrophil count greater than the upper limit of normal). This study included patients regardless of their tumour PD-L1 status. Patients with Karnofsky performance status <70% and patients with any history of or concurrent brain metastases, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. Patients were stratified by IMDC prognostic score and region.
A total of 1096 patients were randomised in the trial, of which 847 patients had intermediate/poor-risk RCC and received either nivolumab 3 mg/kg (n=425) administered intravenously over 60 minutes in combination with ipilimumab 1 mg/kg administered intravenously over 30 minutes every 3 weeks for 4 doses followed by nivolumab monotherapy 3 mg/kg every 2 weeks or sunitinib (n=422) 50 mg daily, administered orally for 4 weeks followed by 2 weeks off, every cycle. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. The first tumour assessments were conducted 12 weeks after randomisation and continued every 6 weeks thereafter for the first year and then every 12 weeks until progression or treatment discontinuation, whichever occurred later. Treatment beyond initial investigator-assessed RECIST, version 1.1-defined progression was permitted if the patient had a clinical benefit and was tolerating study drug as determined by the investigator. The primary efficacy outcome measures were OS, ORR and PFS as determined by a BICR in intermediate/poor risk patients.
Baseline characteristics were generally balanced between the two groups. The median age was 61 years (range: 21-85) with 38% ≥65 years of age and 8% ≥75 years of age. The majority of patients were male (73%) and white (87%), and 31% and 69% of patients had a baseline KPS of 70 to 80% and 90 to 100%, respectively. The median duration of time from initial diagnosis to randomisation was 0.4 years in both the nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg and sunitinib groups. The median duration of treatment was 7.9 months (range: 1 day - 21.4+ months) in nivolumab with ipilimumab-treated patients and was 7.8 months (range: 1 days - 20.2+ months) in sunitinib-treated patients. Nivolumab with ipilimumab was continued beyond progression in 29% of patients.
Efficacy results for the intermediate/poor risk patients are shown in Table 25 (primary analysis with a minimum follow-up of 17.5 months and with a minimum follow-up of 60 months) and in Figure 15 (minimum follow-up of 60 months).
OS results at an additional descriptive analysis undertaken at a minimum follow-up of 60 months show outcomes consistent with the original primary analysis.
Table 25. Efficacy results in intermediate/poor risk patients (CA209214):
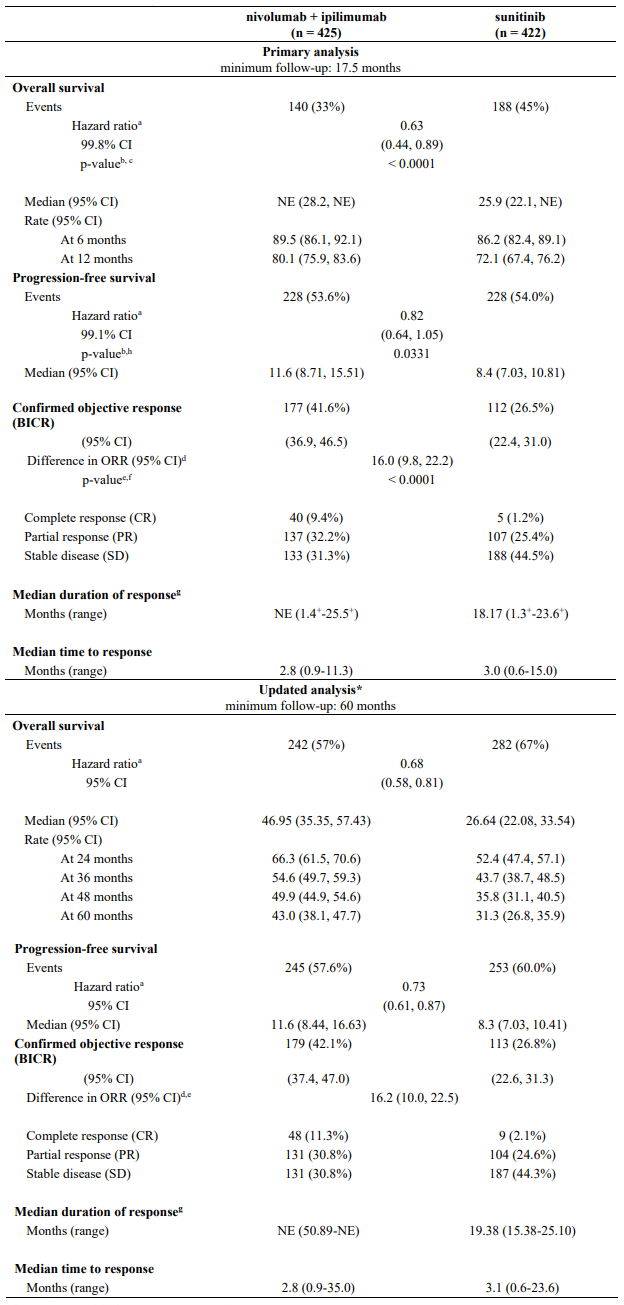
Figure 15. Kaplan-Meier curves of OS in intermediate/poor risk patients (CA209214) - Minimum follow-up of 60 months:
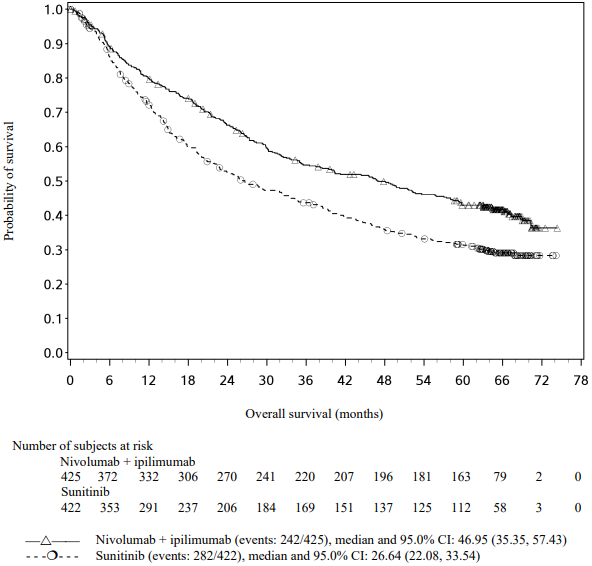
An updated descriptive OS analysis was performed when all patients had a minimum follow-up of 24 months. At the time of this analysis, the hazard ratio was 0.66 (99.8% CI 0.48-0.91) with 166/425 events in the combination arm and 209/422 events in the sunitinib arm. In intermediate/poor-risk patients, OS benefit was observed in the nivolumab in combination with ipilimumab arm vs. sunitinib regardless of tumour PD-L1 expression. Median OS for tumour PD-L1 expression ≥1% was not reached for nivolumab in combination with ipilimumab, and was 19.61 months in the sunitinib arm (HR = 0.52; 95% CI: 0.34, 0.78). For tumour PD-L1 expression <1%, the median OS was 34.7 months for the nivolumab in combination with ipilimumab, and was 32.2 months in the sunitinib arm (HR = 0.70; 95% CI: 0.54, 0.92).
CA209214 also randomised 249 favourable risk patients as per IMDC criteria to nivolumab plus ipilimumab (n=125) or to sunitinib (n=124). These patients were not evaluated as part of the primary efficacy population. At a minimum of 24 months follow-up, OS in favourable risk patients receiving nivolumab plus ipilimumab compared to sunitinib had a hazard ratio of 1.13 (95% CI: 0.64, 1.99; p=0.6710). With 60 months minimum follow-up, the HR for OS was 0.94 (95% CI: 0.65, 1.37).
There are no data on the use of nivolumab in combination with ipilimumab in patients with only a non clear-cell histology in first-line RCC.
Patients ≥75 years of age represented 8% of all intermediate/poor risk patients in CA209214, and the combination of nivolumab and ipilimumab showed numerically less effect on OS (HR 0.97, 95% CI: 0.48, 1.95) in this subgroup versus the overall population at a minimum follow-up of 17.5 months. Because of the small size of this subgroup, no definitive conclusions can be drawn from these data.
Intravenous formulation
Randomised phase 3 study of nivolumab in combination with cabozantinib vs. sunitinib (CA2099ER)
The safety and efficacy of nivolumab 240 mg in combination with cabozantinib 40 mg for the first-line treatment of advanced/metastatic RCC was evaluated in a phase 3, randomised, open-label study (CA2099ER). The study included patients (18 years or older) with advanced or metastatic RCC with a clear cell component, Karnofsky Performance Status (KPS) ≥70%, and measurable disease as per RECIST v1.1 regardless of their PD-L1 status or IMDC risk group. The study excluded patients with autoimmune disease or other medical conditions requiring systemic immunosuppression, patients who had prior treatment with an anti-PD-1, anti PD-L1, anti-PD-L2, anti-CD137, or anti-CTLA-4 antibody, poorly controlled hypertension despite antihypertensive therapy, active brain metastases and uncontrolled adrenal insufficiency. Patients were stratified by IMDC prognostic score, PD-L1 tumour expression, and region.
A total of 651 patients were randomised to receive either nivolumab 240 mg (n=323) administered intravenously every 2 weeks in combination with cabozantinib 40 mg once daily orally or sunitinib (n=328) 50 mg daily, administered orally for 4 weeks followed by 2 weeks off. Treatment continued until disease progression or unacceptable toxicity with nivolumab administration for up to 24 months. Treatment beyond initial investigator-assessed RECIST version 1.1-defined progression was permitted if the patient had a clinical benefit and was tolerating study drug, as determined by the investigator. First tumour assessment post-baseline was performed at 12 weeks (± 7 days) following randomisation. Subsequent tumour assessments occurred at every 6 weeks (± 7 days) until Week 60, then every 12 weeks (± 14 days) until radiographic progression, confirmed by the BICR. The primary efficacy outcome measure was PFS as determined by a BICR. Additional efficacy measures included OS and ORR as key secondary endpoints.
Baseline characteristics were generally balanced between the two groups. The median age was 61 years (range: 28-90) with 38.4% ≥65 years of age and 9.5% ≥75 years of age. The majority of patients were male (73.9%) and white (81.9%). Eight percent of patients were Asian, 23.2% and 76.5% of patients had a baseline KPS of 70 to 80% and 90 to 100%, respectively. Patient distribution by IMDC risk categories was 22.6% favourable, 57.6% intermediate, and 19.7% poor. For tumour PD-L1 expression, 72.5% of patients had PD-L1 expression <1% or indeterminate and 24.9% of patients had PD-L1 expression ≥1%. 11.5% of patients had tumours with sarcomatoid features. The median duration of treatment was 14.26 months (range: 0.2-27.3 months) in nivolumab with cabozantinib-treated patients and was 9.23 months (range: 0.8-27.6 months) in sunitinib-treated patients.
The study demonstrated a statistically significant benefit in PFS, OS, and ORR for patients randomised to nivolumab in combination with cabozantinib as compared to sunitinib. Efficacy results from the primary analysis (minimum follow-up 10.6 months; median follow-up 18.1 months) are shown in Table 26.
Table 26. Efficacy results (CA2099ER):
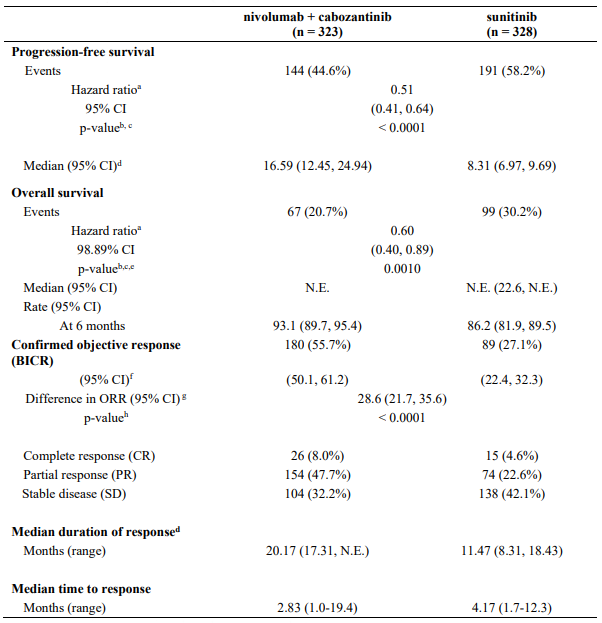
a Stratified Cox proportional hazards model. Hazard ratio is nivolumab and cabozantinib over sunitinib.
b Log-rank test stratified by IMDC prognostic risk score (0, 1-2, 3-6), PD-L1 tumour expression (≥1% versus <1% or indeterminate) and region (US/Canada/W Europe/N Europe, ROW) as entered in the IRT.
c 2-sided p-values from stratified regular log-rank test.
d Based on Kaplan-Meier estimates. e Boundary for statistical significance p-value <0.0111.
f CI based on the Clopper and Pearson method.
g Strata adjusted difference in objective response rate (nivolumab + cabozantinib - sunitinib) based on DerSimonian and Laird.
h 2-sided p-value from CMH test.
NE = non-estimable
The primary analysis of PFS included censoring for new anti-cancer treatment (Table 26). Results for PFS with and without censoring for new anti-cancer treatment were consistent.
PFS benefit was observed in the nivolumab in combination with cabozantinib arm vs. sunitinib regardless of the IMDC risk category. Median PFS for the favourable risk group was not reached for nivolumab in combination with cabozantinib, and was 12.81 months in the sunitinib arm (HR = 0.60; 95% CI: 0.37, 0.98). Median PFS for the intermediate risk group was 17.71 months for nivolumab in combination with cabozantinib and was 8.38 months in the sunitinib arm (HR = 0.54; 95% CI: 0.41, 0.73). Median PFS for the poor risk group was 12.29 months for nivolumab in combination with cabozantinib and was 4.21 months in the sunitinib arm (HR = 0.36; 95% CI: 0.23, 0.58).
PFS benefit was observed in the nivolumab in combination with cabozantinib arm vs. sunitinib regardless of tumour PD-L1 expression. Median PFS for tumour PD-L1 expression ≥1% was 13.08 months for nivolumab in combination with cabozantinib, and was 4.67 months in the sunitinib arm (HR = 0.45; 95% CI: 0.29, 0.68). For tumour PD-L1 expression <1%, the median PFS was 19.84 months for nivolumab in combination with cabozantinib, and 9.26 months in the sunitinib arm (HR = 0.50; 95% CI: 0.38, 0.65).
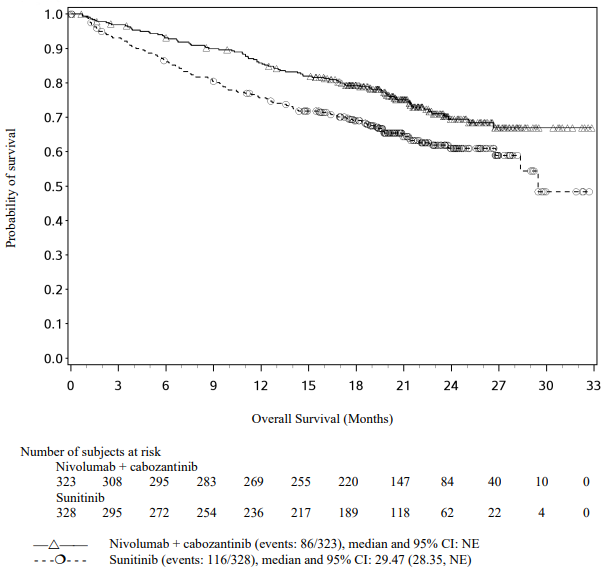
An updated PFS and OS analysis were performed when all patients had a minimum follow-up of 16.0 months and a median follow-up of 23.5 months (see Figures 16 and 17). The PFS hazard ratio was 0.52 (95% CI: 0.43, 0.64). The OS hazard ratio was 0.66 (95% CI: 0.50, 0.87). Updated efficacy data (PFS and OS) in subgroups for the IMDC risk categories and PD-L1 expression levels confirmed the original results. With the updated analysis, median PFS is reached for the favourable risk group.
Figure 16. Kaplan-Meier curves of PFS (CA2099ER):
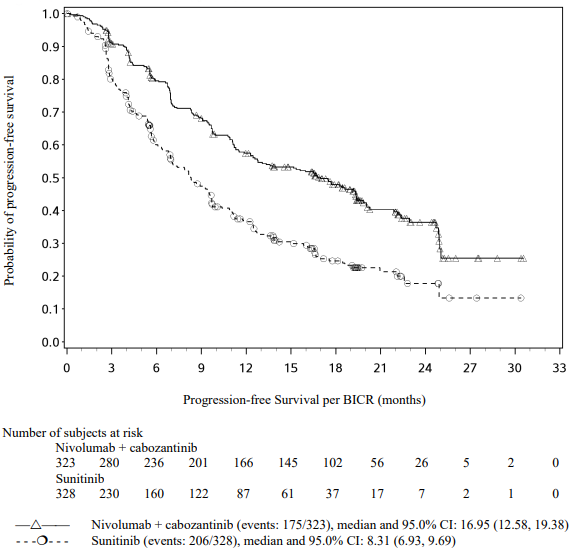
Figure 17. Kaplan-Meier curves of OS (CA2099ER):
Subcutaneous formulation
Randomised, open-label phase 3 study vs. intravenous nivolumab (CA20967T)
The safety and efficacy of nivolumab subcutaneous formulation was evaluated in a multicentre, randomised, open-label study in patients with advanced or metastatic clear cell RCC (CA20967T). Patients 18 years of age or older with histologically confirmed advanced or metastatic RCC with a clear cell component, including those with sarcomatoid features, and who received no more than 2 prior systemic treatment regimens were randomised to receive nivolumab 1200 mg every 4 weeks subcutaneously or nivolumab 3 mg/kg every 2 weeks intravenously. Patients with untreated, symptomatic CNS metastases; leptomeningeal metastases; concurrent malignancies requiring treatment or history of prior malignancy within the prior 2 years; active, known, or suspected autoimmune disease; or who received prior treatment with a checkpoint inhibitor were excluded from the study. Patients with asymptomatic, stable CNS metastases that did not require immediate treatment were eligible if there was no evidence of progression within 28 days prior to the first dose of study drug administration.
Stratification factors for randomisation were weight (<80 kg vs ≥80 kg) and International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) risk classification (favourable vs intermediate vs poor risk).
The primary objective of the study was to demonstrate noninferiority of the serum nivolumab Cavgd28 and Cminss for the subcutaneous administration of nivolumab to the intravenous administration of nivolumab (see section 5.2). The key secondary objective of the study was to demonstrate noninferiority of the ORR for the subcutaneous administration of nivolumab to the intravenous administration of nivolumab, as assessed by blinded independent central review (BICR). Additional secondary objectives included assessing duration of response (DOR), progression-free survival (PFS), and overall survival (OS).
A total of 495 patients were randomised to receive either subcutaneous nivolumab (n=248) or intravenous nivolumab (n=247). The median age was 65 years (range: 20 to 93), with 51% ≥65 years of age and 14% ≥75 years of age, 85% White, 0.8% Asian, and 0.4% Black, and 68% male. Fifty-seven percent of patients weighed <80 kg and 43% weighed ≥80 kg. Baseline Karnofsky performance status was 70 (7%), 80 (20%), 90 (34%), or 100 (39%). Patient distribution by IMDC risk categories was 21% favourable, 62% intermediate, and 17% poor.
The study demonstrated noninferiority of nivolumab 1200 mg administered subcutaneously to nivolumab 3 mg/kg administered intravenously (see section 5.2). At the primary analysis (minimum follow-up of 8 months) ORR was 24.2% (95% CI: 19.0, 30.0) for subcutaneous nivolumab and 18.2% (95% CI: 13.6, 23.6) for intravenous nivolumab. The estimate of objective response risk ratio was 1.33 (95% CI: 0.94, 1.88). To declare noninferiority, the lower bound of the two-sided 95% CI of the objective response risk ratio had to be ≥0.60. Updated efficacy results with a minimum follow-up of 14.9 months (data cut-off 21-Feb-2024) are shown in Table 27.
Table 27. Efficacy results - CA20967T:
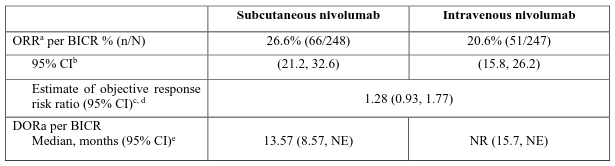
NR = not reached, NE = non-estimable
a Descriptive analysis.
b Confidence interval based on the Clopper and Pearson method.
c Stratified by weight (<80 kg vs ≥80 kg) and IMDC risk group (favourable vs intermediate vs poor).
d Strata adjusted risk ratio (subcutaneous nivolumab over intravenous nivolumab) using Mantel-Haenszel method.
e Median computed using Kaplan-Meier method.
Intravenous formulation
Randomised phase 3 study of nivolumab as monotherapy vs. everolimus (CA209025)
The safety and efficacy of nivolumab 3 mg/kg as a single agent for the treatment of advanced RCC with a clear cell component was evaluated in a Phase 3, randomised, open-label study (CA209025). The study included patients (18 years or older) who have experienced disease progression during or after 1 or 2 prior anti-angiogenic therapy regimens and no more than 3 total prior systemic treatment regimens. Patients had to have a Karnofsky Performance Score (KPS) ≥70%. This study included patients regardless of their tumour PD-L1 status. Patients with any history of or concurrent brain metastases, prior treatment with an mammalian target of rapamycin (mTOR) inhibitor, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study.
A total of 821 patients were randomised to receive either nivolumab 3 mg/kg (n=410) administered intravenously over 60 minutes every 2 weeks or everolimus (n=411) 10 mg daily, administered orally. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. The first tumour assessments were conducted 8 weeks after randomisation and continued every 8 weeks thereafter for the first year and then every 12 weeks until progression or treatment discontinuation, whichever occurred later. Tumour assessments were continued after treatment discontinuation in patients who discontinued treatment for reasons other than progression. Treatment beyond initial investigator-assessed RECIST, version 1.1-defined progression was permitted if the patient had a clinical benefit and was tolerating study drug as determined by the investigator. The primary efficacy outcome measure was OS. Secondary efficacy assessments included investigator-assessed ORR and PFS.
Baseline characteristics were generally balanced between the two groups. The median age was 62 years (range: 18-88) with 40% ≥65 years of age and 9% ≥75 years of age. The majority of patients were male (75%) and white (88%), all Memorial Sloan Kettering Cancer Center (MSKCC) risk groups were represented, and 34% and 66% of patients had a baseline KPS of 70 to 80% and 90 to 100%, respectively. The majority of patients (72%) were treated with one prior anti-angiogenic therapy. The median duration of time from initial diagnosis to randomisation was 2.6 years in both the nivolumab and everolimus groups. The median duration of treatment was 5.5 months (range: 0-29.6+ months) in nivolumab-treated patients and was 3.7 months (range: 6 days-25.7+ months) in everolimus-treated patients.
Nivolumab was continued beyond progression in 44% of patients.
The Kaplan-Meier curves for OS are shown in Figure 18.
Figure 18. Kaplan-Meier curves of OS (CA209025):
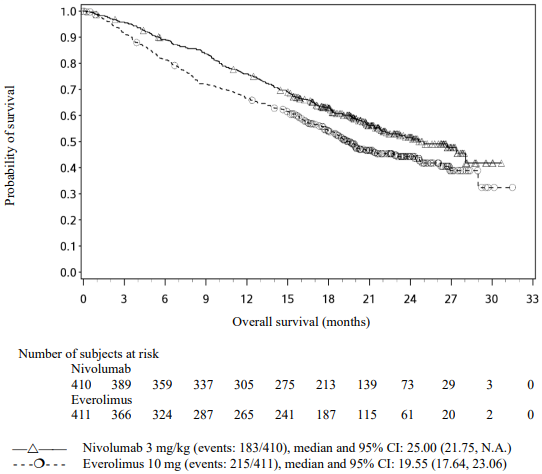
The trial demonstrated a statistically significant improvement in OS for patients randomised to nivolumab as compared with everolimus at the prespecified interim analysis when 398 events were observed (70% of the planned number of events for final analysis) (Table 38 and Figure 22). OS benefit was observed regardless of tumour PD-L1 expression level.
Efficacy results are shown in Table 28.
Table 28. Efficacy results (CA209025):
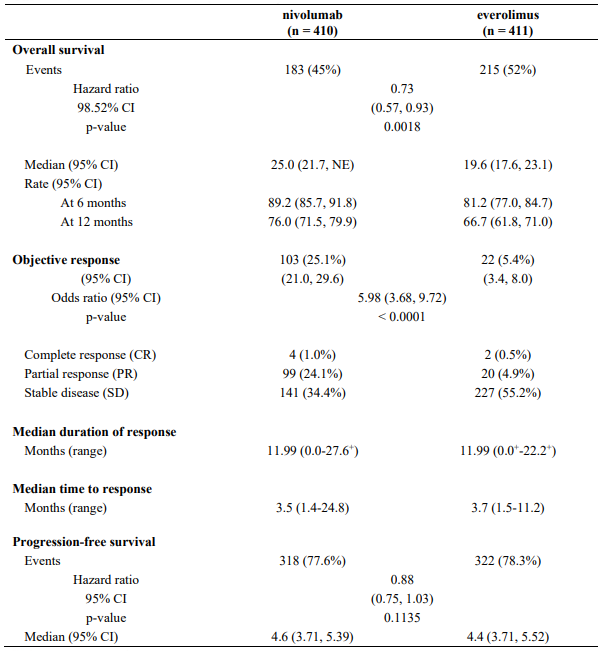
"+" denotes a censored observation.
NE = non-estimable
The median time to onset of objective response was 3.5 months (range: 1.4-24.8 months) after the start of nivolumab treatment. Forty-nine (47.6%) responders had ongoing responses with a duration ranging from 0.0-27.6+ months.
Overall survival could be accompanied by an improvement over time in disease related symptoms and non-disease specific QoL as assessed using valid and reliable scales in the Functional Assessment of Cancer Therapy-Kidney Symptom Index-Disease Related Symptoms (FKSI-DRS) and the EuroQoL EQ-5D. Apparently meaningful symptom improvement (MID = 2 point change in FKSI-DRS score; p<0.001) and time to improvement (HR = 1.66 (1.33, 2.08), p<0.001) were significantly better for patients on the nivolumab arm. While both arms of the study received active therapy, the QoL data should be interpreted in the context of the open-label study design and therefore cautiously taken.
Intravenous formulation
Phase 3b/4 safety study (CA209374)
Additional safety and descriptive efficacy data are available from study CA209374, an open-label Phase 3b/4 safety study of nivolumab monotherapy (treated with 240 mg every 2 weeks) for the treatment of patients with advanced or metastatic RCC (n=142), including 44 patients with non-clear cell histology.
In subjects with non-clear cell histology, at a minimum follow-up of approximately 16.7 months ORR and median duration of response were 13.6% and 10.2 months, respectively. Clinical activity was observed regardless of tumour PD-L1 expression status.
Squamous cell cancer of the head and neck
Intravenous formulation
The safety and efficacy of nivolumab 3 mg/kg as a single agent for the treatment of metastatic or recurrent SCCHN were evaluated in a phase 3, randomised, open-label study (CA209141). The study included patients (18 years or older), with histologically confirmed recurrent or metastatic SCCHN (oral cavity, pharynx, larynx), stage III/IV and not amenable to local therapy with curative intent (surgery or radiation therapy with or without chemotherapy) and who have experienced disease progression during or within 6 months of receiving platinum-based therapy regimen and had an ECOG performance status score of 0 or 1. Prior platinum-based therapy was administered in either the adjuvant, neo-adjuvant, primary, recurrent, or metastatic setting. Patients were enrolled regardless of their tumour PD-L1 or human papilloma virus (HPV) status. Patients with active autoimmune disease, medical conditions requiring immunosuppression, recurrent or metastatic carcinoma of the nasopharynx, squamous cell carcinoma of unknown primary, salivary gland or non-squamous histologies (e.g., mucosal melanoma), or active brain or leptomeningeal metastases were excluded from the study. Patients with treated brain metastases were eligible if neurologically returned to baseline at least 2 weeks prior to enrolment, and either off corticosteroids, or on a stable or decreasing dose of <10 mg daily prednisone equivalents.
A total of 361 patients were randomised to receive either nivolumab 3 mg/kg (n=240) administered intravenously over 60 minutes every 2 weeks or investigator's choice of either cetuximab (n=15), 400 mg/m² loading dose followed by 250 mg/m² weekly or methotrexate (n=52) 40 to 60 mg/m² weekly, or docetaxel (n=54) 30 to 40 mg/m² weekly. Randomisation was stratified by prior cetuximab treatment. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. Tumour assessments, according to RECIST version 1.1, were conducted 9 weeks after randomisation and continued every 6 weeks thereafter. Treatment beyond initial investigator-assessed RECIST version 1.1-defined progression was permitted in patients receiving nivolumab, if the patient had a clinical benefit and was tolerating study drug, as determined by the investigator. The primary efficacy outcome measure was OS. Key secondary efficacy outcome measures were investigator-assessed PFS and ORR. Additional prespecified subgroup analyses were conducted to evaluate the efficacy by tumour PD-L1 expression at predefined levels of 1%, 5%, and 10%.
Pre-study tumour tissue specimens were systematically collected prior to randomisation in order to conduct pre-planned analyses of efficacy according to tumour PD-L1 expression. Tumour PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay.
Baseline characteristics were generally balanced between the two groups. The median age was 60 years (range: 28-83) with 31% ≥65 years of age and 5% ≥75 years of age, 83% were male, and 83% were white. Baseline ECOG performance status score was 0 (20%) or 1 (78%), 77% were former/current smokers, 90% had Stage IV disease, 66% had two or more lesions, 45%, 34% and 20% received 1, 2, or 3 or more prior lines of systemic therapy, respectively, and 25% were HPV-16 status positive.
With a minimum follow-up of 11.4 months, the trial demonstrated a statistically significant improvement in OS for patients randomised to nivolumab as compared with investigator's choice. The Kaplan-Meier curves for OS are shown in Figure 19. Efficacy results are shown in Table 29.
Figure 19. Kaplan-Meier curves of OS (CA209141):
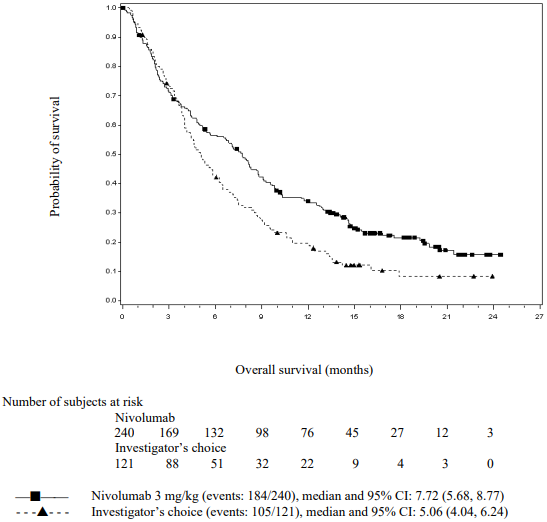
Table 29. Efficacy results (CA209141):
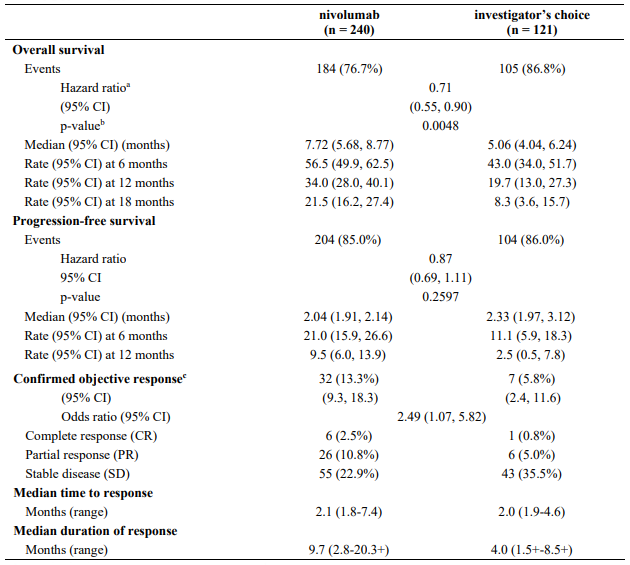
a Derived from a stratified proportional hazards model.
b P-value is derived from a log-rank test stratified by prior cetuximab; the corresponding O'Brien-Fleming efficacy boundary significance level is 0.0227.
c In the nivolumab group there were two patients with CRs and seven patients with PRs who had tumour PD-L1 expression <1%.
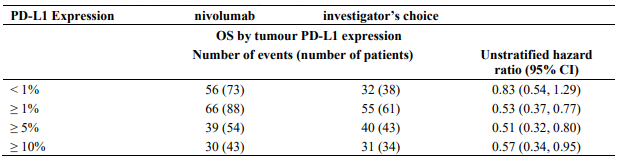
Quantifiable tumour PD-L1 expression was measured in 67% of patients in the nivolumab group and 82% of patients in the investigator's choice group. Tumour PD-L1 expression levels were balanced between the two treatment groups (nivolumab vs. investigator's choice) at each of the predefined tumour PD-L1 expression levels of ≥1% (55% vs. 62%), ≥5% (34% vs. 43%), or ≥10% (27% vs. 34%).
Patients with tumour PD-L1 expression by all predefined expression levels in the nivolumab group demonstrated greater likelihood of improved survival compared to investigator's choice. The magnitude of OS benefit was consistent for ≥1%, ≥5% or ≥10% tumour PD-L1 expression levels (see Table 30).
Table 30. OS by tumour PD-L1 expression (CA209141):
In an exploratory post-hoc analysis using a non-validated assay, both tumour cell PD-L1 expression and tumour-associated immune cell (TAIC) PD-L1 expression were analysed in relation to the magnitude of treatment effect of nivolumab compared to investigator's choice. This analysis showed that not only tumour cell PD-L1 expression but also TAIC PD-L1 expression appeared to be associated with benefit from nivolumab relative to investigator's choice (see Table 44). Due to the small numbers of patients in the subgroups, and exploratory nature of the analysis, no definitive conclusions can be drawn from these data.
Table 31. Efficacy by tumour cell and TAIC PD-L1 expression (CA209141):
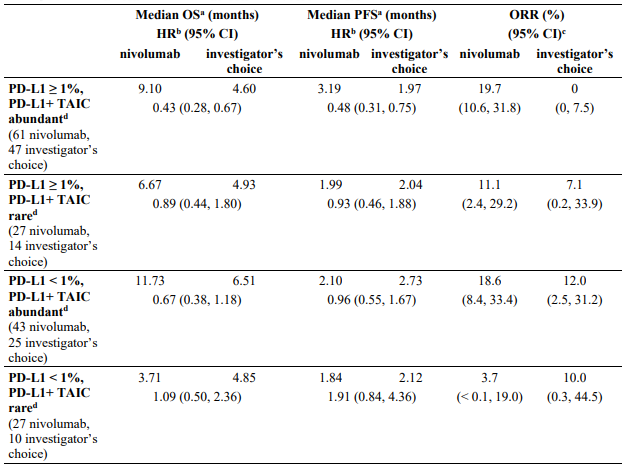
a OS and PFS were estimated using Kaplan-Meier method.
b Hazard ratio in each subgroup derived from a Cox proportional hazards model with treatment as the only covariate.
c Confidence interval for ORR calculated using the Clopper-Pearson method.
d PD-L1+ TAIC in the tumour microenvironment were qualitatively assessed, and characterised as "numerous", "intermediate", and "rare" based on pathologist assessments. "Numerous" and "intermediate" groups were combined to define the "abundant" group.
Patients with investigator-assessed primary site of oropharyngeal cancer were tested for HPV (determined by p16 immunohistochemistry [IHC]). OS benefit was observed regardless of HPV status (HPV-positive: HR = 0.63; 95% CI: 0.38, 1.04, HPV-negative: HR = 0.64; 95% CI: 0.40, 1.03, and HPV-unknown: HR = 0.78; 95% CI: 0.55, 1.10).
Patient-reported outcomes (PROs) were assessed using the EORTC QLQ-C30, EORTC QLQ-H&N35, and 3-level EQ-5D. Over 15 weeks of follow-up, patients treated with nivolumab exhibited stable PROs, while those assigned to investigator's choice therapy exhibited significant declines in functioning (e.g., physical, role, social) and health status as well as increased symptomatology (e.g., fatigue, dyspnoea, appetite loss, pain, sensory problems, social contact problems). The PRO data should be interpreted in the context of the open-label study design and therefore taken cautiously.
Urothelial carcinoma
Adjuvant treatment of urothelial carcinoma
Intravenous formulation
Randomised phase 3 study of adjuvant nivolumab vs. placebo (CA209274):
The safety and efficacy of nivolumab monotherapy for the adjuvant treatment of urothelial carcinoma was evaluated in a phase 3 multicentre, randomised, placebo-controlled, double-blinded study (CA209274). The study included patients (18 years or older) who have undergone radical resection of muscle invasive urothelial carcinoma (MIUC) originating in the bladder or upper urinary tract (renal pelvis or ureter) and are at high risk of recurrence. The MIUC pathologic staging criteria that defines high risk patients was ypT2-ypT4a or ypN+ for adult patients who received neoadjuvant cisplatin chemotherapy, and pT3-pT4a or pN+ for adult patients who did not receive neoadjuvant cisplatin chemotherapy and were not eligible or refused adjuvant cisplatin chemotherapy. The study included patients regardless of their PD-L1 status, who had an ECOG performance status score of 0 or 1 (an ECOG PS of 2 was allowed for patients ineligible for neoadjuvant cisplatin chemotherapy). Tumour cell PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay. The study excluded patients with active, known or suspected autoimmune disease, patients who had treatment with any chemotherapy, radiation therapy, biologics for cancer, intravesical therapy, or investigational therapy within 28 days of first administration of study treatment.
A total of 709 patients were randomised to receive either nivolumab 240 mg (n=353) every 2 weeks or placebo (n=356) every 2 weeks until recurrence or unacceptable toxicity for a maximum treatment duration of 1 year. Of these, 282 patients had tumour cell PD-L1 expression ≥1%; 140 in the nivolumab arm and 142 in the placebo arm. Randomisation was stratified by pathologic nodal status (N+ vs. N0/x with <10 nodes removed vs. N0 with ≥10 nodes removed), tumour cell PD-L1 expression (≥1% vs. <1%/indeterminate), and use of cisplatin neoadjuvant chemotherapy. Tumour imaging assessments were to be performed every 12 weeks from the date of first dose to week 96, then every 16 weeks from week 96 to week 160, then every 24 weeks until non-urothelial tract recurrence or treatment was discontinued (whichever occurred later) for a maximum of 5 years. The primary efficacy outcome measures were disease-free survival (DFS) in all randomised patients and DFS in randomised patients with tumour cell PD-L1 expression ≥1%. DFS was defined as the time between the date of randomisation and the date of the first documented recurrence assessed by investigator (local urothelial tract, local non-urothelial tract or distant), or death (from any cause), whichever occurred first. Secondary efficacy outcome measures included overall survival (OS).
Baseline characteristics were generally balanced across treatment groups. In patients with tumour cell PD-L1 expression ≥1%, the median age was 66 years (range: 34-92 years), 76% were male and 76% were white. Eighty two percent had muscle invasive bladder cancer (MIBC), 18% had upper tract urothelial carcinoma (UTUC) (renal pelvis and ureter), 42% of patients received prior cisplatin in the neoadjuvant setting, 45% of patients were N+ at radical resection, patients had ECOG performance status of 0 (61%), 1 (37%), or 2 (2%), and 7% of patients had a haemoglobin <10 g/dL.
At the primary pre-specified interim analysis in patients with tumour cell PD-L1 expression ≥1% (minimum follow-up of 6.3 months and median follow-up of 22.1 months for the nivolumab arm), the study demonstrated a statistically significant improvement in DFS for patients randomised to nivolumab as compared to placebo. Median DFS as determined by the investigator was not reached (95% CI: 21.19, N.R.) for nivolumab versus 8.41 months (95% CI: 5.59, 21.19) for placebo, HR 0.55 (98.72% CI: 0.35, 0.85), p-value = 0.0005. The primary analysis of DFS included censoring for new anti-cancer treatment. Results for DFS with and without censoring for new anti-cancer treatment were consistent.
In an updated descriptive DFS analysis in patients with tumour cell PD-L1 expression ≥1% (minimum follow-up of 11.4 months and median follow-up of 25.5 months for the nivolumab arm), DFS improvement was confirmed.
Efficacy results from this descriptive updated analysis are shown in Table 32 and Figure 20.
Table 32. Efficacy results in patients with tumour cell PD-L1 ≥1% (CA209274):
NR: not reached, NE: non-estimable.
a Stratified Cox proportional hazard model. Hazard Ratio is nivolumab over placebo.
b Based on Kaplan-Meier estimates.
Figure 20. Kaplan-Meier curves of DFS in patients with tumour cell PD-L1 expression ≥1% (CA209274):
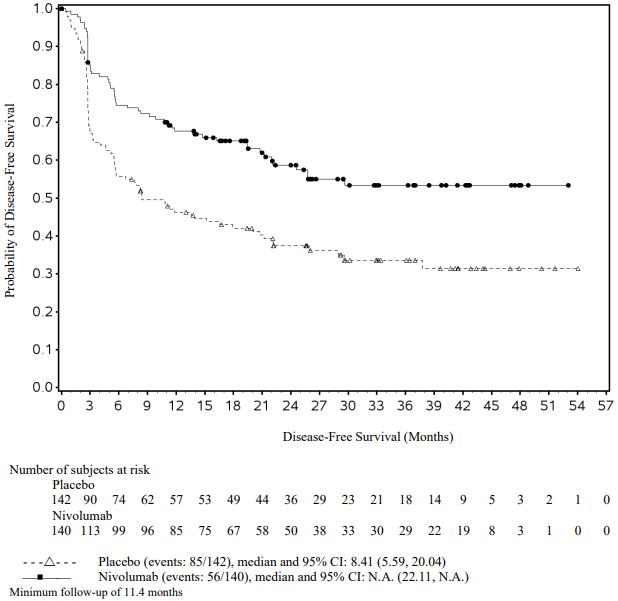
Exploratory pre-specified subgroup descriptive analyses were performed in patients based on prior cisplatin treatment in the neoadjuvant setting.
In the subgroup of patients with tumour cell PD-L1 expression ≥1% who received prior cisplatin in the neoadjuvant setting (n=118), the DFS HR was 0.37 (95% CI: 0.22, 0.64) with median DFS not reached and 8.41 months for the nivolumab and placebo arms, respectively. In the subgroup of patients with tumour cell PD-L1 expression ≥1% who did not receive prior cisplatin in the neoadjuvant setting (n=164), the DFS HR was 0.69 (95% CI: 0.44, 1.08) with median DFS of 29.67 and 11.37 months for the nivolumab and placebo arms, respectively.
Treatment of advanced urothelial carcinoma
Intravenous formulation
Randomised open-label phase 3 study of nivolumab in combination with chemotherapy vs. chemotherapy (CA209901)
The safety and efficacy of nivolumab in combination with cisplatin and gemcitabine followed by nivolumab monotherapy were evaluated in a randomised open-label study CA209901 in cisplatin-eligible patients with unresectable or metastatic urothelial carcinoma. The study included subjects (18 years or older) with histological or cytological evidence of metastatic or surgically unresectable transitional cell carcinoma (TCC) of the urothelium involving the renal pelvis, ureter, bladder or urethra, who were eligible for cisplatin and gemcitabine. Minor histologic variants (<50% overall) were acceptable (TCC must have been the dominant histology). All subjects were required to have measurable disease by computed tomography (CT) or magnetic resonance imaging (MRI) per RECIST 1.1 criteria. No prior systemic anti-cancer therapy for metastatic or surgically unresectable urothelial carcinoma was permitted. Prior neoadjuvant chemotherapy or prior adjuvant platinum-based chemotherapy following radical cystectomy were permitted as long as the disease recurrence took place ≥12 months from completion of therapy. Prior intravesical therapy was permitted if completed at least 4 weeks prior to initiation of study treatment. Radiation therapy (with or without chemotherapy) with curative intent was permitted if treatment was completed ≥12 months before enrolment. Palliative radiotherapy was permitted as long as it was completed at least 2 weeks prior to therapy.
A total of 608 patients were randomised to receive either nivolumab in combination with cisplatin and gemcitabine (n=304) or cisplatin and gemcitabine (n=304). Randomisation was stratified by tumour PD-L1 status (≥1% vs. <1% or indeterminate) and liver metastasis (yes vs. no). The median age was 65 years of age (range: 32 to 86) with 51% of patients ≥65 years of age and 12% of patients ≥75 years of age, 23% were Asian, 72% were White, 0.3% were Black; 77% were male, 23% were female. Baseline ECOG performance status was 0 (53%) or 1 (46%). Patients in the nivolumab in combination with cisplatin and gemcitabine arm were treated with nivolumab 360 mg every three weeks, in combination with cisplatin and gemcitabine for up to 6 cycles, after which patients received nivolumab monotherapy 480 mg every 4 weeks for a total of up to 24 months. Patients received gemcitabine dosed at 1000 mg/m² IV over 30-minutes on Days 1 and 8 of the 3 week treatment cycle and cisplatin dosed at 70 mg/m² IV over 30 to 120-minutes on Day 1 of the 3 week treatment cycle. A total of 92 patients (49 in the nivolumab in combination with cisplatin and gemcitabine arm and 43 in the cisplatin and gemcitabine arm) switched from cisplatin to carboplatin after at least one cycle of cisplatin.
The study demonstrated a statistically significant benefit in OS and PFS for patients randomised to nivolumab in combination with cisplatin and gemcitabine compared to cisplatin and gemcitabine alone. Efficacy results are presented in Table 33 and Figures 21 and 22.
Table 33. Efficacy Results (CA209901):
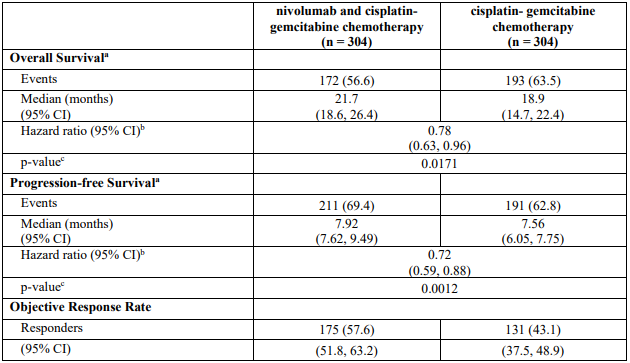
a Based on Kaplan-Meier Estimates
b Stratified Cox proportional hazard model.
c 2 sided p-value from stratified log-rank test.
Figure 21. Kaplan Meier curves of OS (CA209901):
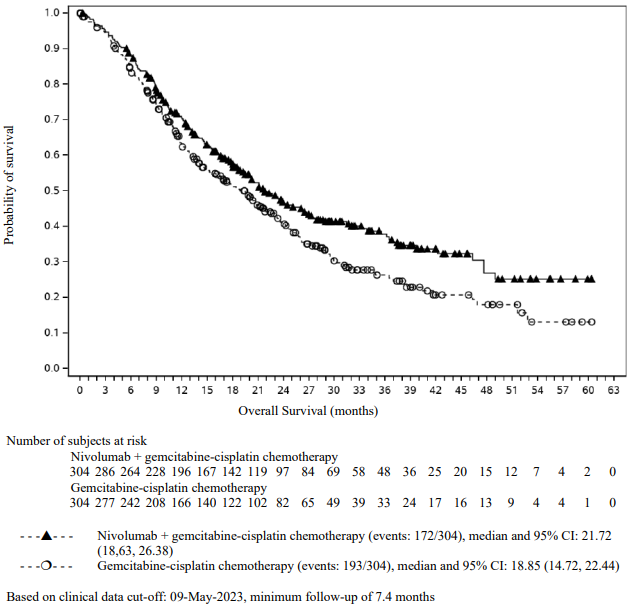
Figure 22. Kaplan Meier curves of PFS (CA209901):
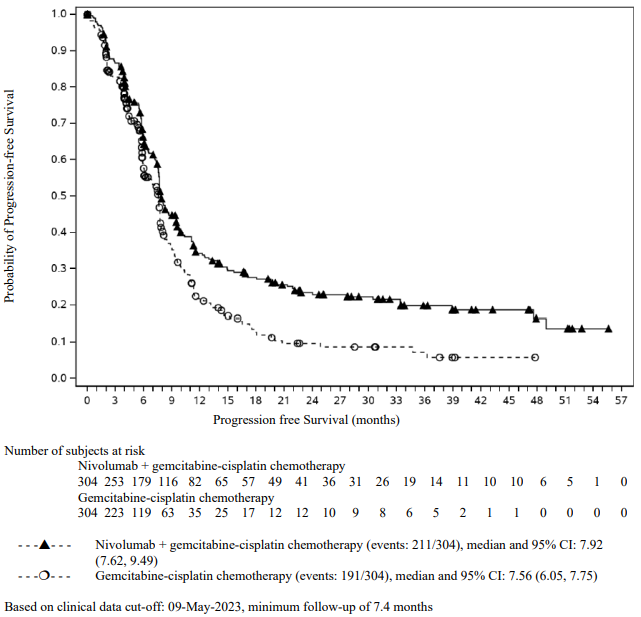
Based on clinical data cut-off: 09-May-2023, minimum follow-up of 7.4 months
The primary analysis of PFS included censoring for new anti-cancer treatment before disease progression (Table 46). Results for PFS with and without censoring for new anti-cancer treatment before disease progression were consistent.
Intravenous formulation
Open-label phase 2 study (CA209275)
The safety and efficacy of nivolumab 3 mg/kg as a single agent for the treatment of patients with locally advanced or metastatic urothelial carcinoma was evaluated in a phase 2, multicentre, open-label, single-arm study (CA209275).
The study included patients (18 years or older) who had disease progression during or following platinum-containing chemotherapy for advanced or metastatic disease or had disease progression within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. Patients had an ECOG performance status score of 0 or 1 and were enrolled regardless of their tumour PD-L1 status. Patients with active brain metastases or leptomeningeal metastases, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. Patients that received more than 2 prior lines of chemotherapy with liver metastases were excluded.
A total of 270 patients who received nivolumab 3 mg/kg administered intravenously over 60 minutes every 2 weeks with a minimum follow-up of 8.3 months were evaluable for efficacy. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. The first tumour assessments were conducted 8 weeks after the start of treatment and continued every 8 weeks thereafter up to 48 weeks, then every 12 weeks until disease progression or treatment discontinuation, whichever occurred later. Tumour assessments were continued after treatment discontinuation in patients who discontinued treatment for reasons other than progression. Treatment beyond initial investigator-assessed RECIST, version 1.1-defined progression was permitted if the patient had a clinical benefit, did not have rapid disease progression, and was tolerating study drug as determined by the investigator. The primary efficacy outcome measure was ORR as determined by BICR. Additional efficacy measures included duration of response, PFS and OS (see Table 34).
The median age was 66 years (range: 38 to 90) with 55% ≥65 years of age and 14% ≥75 years of age. The majority of patients were white (86%) and male (78%). Baseline ECOG performance status was 0 (54%) or 1 (46%).
Table 34. Efficacy results (CA209275)a:
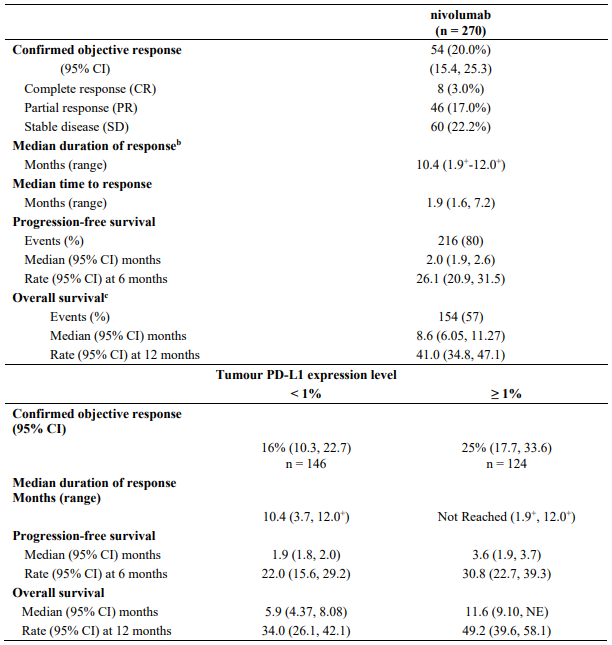
"+" denotes a censored observation.
a median follow-up 11.5 months.
b Data unstable due to the limited duration of response.
c included 4 drug-related deaths: 1 pneumonitis, 1 acute respiratory failure, 1 respiratory failure, and 1 cardiovascular failure.
NE: non-estimable
Results from post-hoc, exploratory analyses indicate that in patients with low (e.g. <1%) to no tumour PD-L1 expression, other patient characteristics (e.g. liver metastases, visceral metastases, baseline haemoglobin <10 g/dL and ECOG performance status = 1) might contribute to the clinical outcome.
Intravenous formulation
Open-label phase ½ study (CA209032)
CA209032 was a Phase ½ open-label multi-cohort study which included a cohort of 78 patients (including 18 subjects who received planned crossover treatment with nivolumab 3 mg/kg plus ipilimumab 1 mg/kg combination) with similar inclusion criteria to study CA209275 treated with nivolumab monotherapy 3 mg/kg for urothelial carcinoma. At a minimum follow-up of 9 months, investigator-assessed confirmed ORR was 24.4% (95% CI: 15.3, 35.4). The median duration of response was not reached (range: 4.4-16.6+ months). The median OS was 9.7 months (95% CI: 7.26, 16.16) and the estimated OS rates were 69.2% (CI: 57.7, 78.2) at 6 months and 45.6% (CI: 34.2, 56.3) at 12 months.
dMMR or MSI-H colorectal cancer
Intravenous formulation
Open-label study of nivolumab in combination with ipilimumab versus chemotherapy in dMMR or MSI-H CRC patients naive to treatment in the metastatic setting
The safety and efficacy of nivolumab 240 mg in combination with ipilimumab 1 mg/kg every 3 weeks, for a maximum of 4 doses, followed by nivolumab monotherapy 480 mg every 4 weeks in the first-line treatment of unresectable or metastatic CRC with known tumour MSI-H or dMMR status were evaluated in a randomised, multi-arm, phase 3, open-label study (CA2098HW). Study treatment arms included nivolumab monotherapy, nivolumab in combination with ipilimumab, or investigator's choice of chemotherapy. MSI-H or dMMR tumour status was determined in accordance with local standard of practice using PCR, NGS or IHC, assays. Central assessment of MSI-H status using PCR (Idylla MSI) test and dMMR status using IHC (Omnis MMR) test was conducted retrospectively on patient tumour specimens used for local MSI-H/dMMR status determination. Patients with confirmed MSI-H/dMMR status by either central test comprised the primary efficacy population. Patients with brain metastasis that were symptomatic, had active autoimmune disease, used systemic corticosteroids or immunosuppressants, or had been treated with checkpoint inhibitors were excluded from the study. Randomisation was stratified by tumour location (right vs left). Patients randomised to the chemotherapy arm could receive nivolumab plus ipilimumab combination upon progression assessed by BICR.
A total of 303 previously untreated patients, in the metastatic setting, were randomised to study, including 202 patients to nivolumab in combination with ipilimumab and 101 patients to chemotherapy. Among them 255 had centrally confirmed MSI-H/dMMR status, 171 in the nivolumab in combination with ipilimumab arm and 84 in the chemotherapy arm. Patients in the nivolumab plus ipilimumab arm received nivolumab 240 mg every 3 weeks in combination with ipilimumab 1 mg/kg every 3 weeks, for a maximum of 4 doses, followed by nivolumab monotherapy 480 mg every 4 weeks. Patients in the chemotherapy arm received: mFOLFOX6 (oxaliplatin, leucovorin, and fluorouracil) with or without either bevacizumab or cetuximab: Oxaliplatin 85 mg/m², leucovorin 400 mg/m², and fluorouracil 400 mg/m² bolus followed by fluorouracil 2400 mg/m² over 46 hours every 2 weeks. Bevacizumab 5 mg/kg or cetuximab 500 mg/m² administered prior to mFOLFOX6 every 2 weeks; or FOLFIRI (irinotecan, leucovorin, and fluorouracil) with or without either bevacizumab or cetuximab: Irinotecan 180 mg/m², leucovorin 400 mg/m², and fluorouracil 400 mg/m² bolus and fluorouracil 2400 mg/m² over 46 hours every 2 weeks. Bevacizumab 5 mg/kg or cetuximab 500 mg/m² administered prior to FOLFIRI every 2 weeks. Treatment continued until disease progression, unacceptable toxicity, or for nivolumab in combination with ipilimumab up to 24 months. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue nivolumab as a single agent. Tumour assessments per RECIST v1.1 were conducted every 6 weeks for the first 24 weeks, then every 8 weeks thereafter until week 96, then every 16 weeks thereafter until week 146, and then every 24 weeks.
The baseline characteristics of all randomised previously untreated for metastatic disease patients were: the median age was 63 years (range: 21 to 87), with 46% ≥65 years of age and 18% ≥75 years of age; 46% were male and 86% were White. Baseline ECOG performance status was 0 (54%) and ≥1 (46%); tumour location was right-sided or left-sided for 68% and 32% of patients, respectively; and 39 patients had confirmed Lynch syndrome among the 223 patients with a known status. The baseline characteristics of previously untreated for metastatic disease patients with centrally confirmed MSI-H/dMMR were consistent with all randomised previously untreated patients. Among the 101 patients randomised to receive chemotherapy, 88 received chemotherapy per protocol, including oxaliplatin-containing regimens (58%) and irinotecan-containing regimens (42%). Additionally, 66 patients received a targeted agent, either bevacizumab (64%) or cetuximab (11%).
A primary efficacy outcome measure of the study was BICR-assessed PFS per RECIST 1.1. Additional efficacy measures included ORR assessed by BICR, OS, and duration of response.
The study met the primary endpoint, at the planned interim analysis, demonstrating a statistically significant improvement in BICR assessed-PFS for patients with centrally confirmed MSI-H/dMMR in the nivolumab in combination with ipilimumab arm compared with the chemotherapy arm. The BICR-assessed PFS results are presented in Table 35 and Figure 23. At the time of this interim analysis, the other endpoints, including the data from nivolumab monotherapy arm, were not tested, due to testing hierarchy.
Table 35. Efficacy results in first-line MSI-H/dMMR centrally confirmed CRC (CA2098HW)a:

a Median follow-up of 31.5 months (range: 6.1 to 48.4 months).
b Based on stratified 2-sided log-rank test
Figure 23. Kaplan-Meier curve of PFS in first-line patients with MSI-H/dMMR centrally confirmed CRC (CA2098HW):
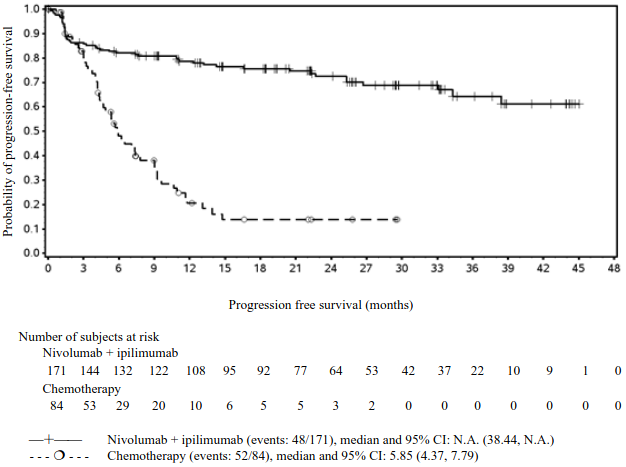
Open-label study of nivolumab in combination with ipilimumab in dMMR or MSI-H CRC in patients who received prior fluoropyrimidine-based combination chemotherapy
The safety and efficacy of nivolumab 3 mg/kg in combination with ipilimumab 1 mg/kg for the treatment of dMMR or MSI-H metastatic CRC was evaluated in a Phase 2, multi-centre, open-label, single-arm study (CA209142).
The study included patients (18 years or older) with locally determined dMMR or MSI-H status, who had disease progression during, after, or were intolerant to, prior therapy with fluoropyrimidine and oxaliplatin or irinotecan. Patients who had their most recent prior treatment in the adjuvant setting should have progressed on or within 6 months of completion of adjuvant chemotherapy. Patients had an ECOG performance status score of 0 or 1 and were enrolled regardless of their tumour PD-L1 status. Patients with active brain metastases, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study.
A total of 119 patients were treated with nivolumab 3 mg/kg administered intravenously over 60 minutes in combination with ipilimumab 1 mg/kg administered intravenously over 90 minutes every 3 weeks for 4 doses followed by nivolumab monotherapy 3 mg/kg every 2 weeks. Treatment was continued as long as clinical benefit was observed or until treatment was no longer tolerated. Tumour assessments according to RECIST version 1.1 were conducted every 6 weeks for the first 24 weeks and every 12 weeks thereafter. The primary outcome measure was investigator-assessed ORR. Secondary outcome measures were BICR-assessed ORR and disease control rate. Analysis of ORR included duration of and time to response. Exploratory outcome measures included PFS and OS.
The median age was 58 years (range: 21-88) with 32% ≥65 years of age and 9% ≥75 years of age, 59% were male and 92% were white. Baseline ECOG performance status was 0 (45%) or 1 (55%), 25% of patients had BRAF mutations, 37% had KRAS mutations, and 12% were unknown. Of the 119 treated patients, 109 had received prior fluoropyrimidine based chemotherapy in the metastatic setting and 9 in the adjuvant setting. Before study enrolment, of the 119 treated patients, 118 (99%) had received fluorouracil, 111 (93%) had received oxaliplatin, 87 (73%) had received irinotecan as part of prior therapies; 82 (69%) had received prior treatment with fluoropyrimidine, oxaliplatin, and irinotecan. Twenty three percent, 36%, 24%, and 16% received 1, 2, 3, or 4 or more prior therapies respectively, and 29% of patients had received an EGFR inhibitor.
Efficacy results (minimum follow-up 46.9 months; median follow-up 51.1 months) are shown in Table 36.
Table 36. Efficacy results (CA209142)*:
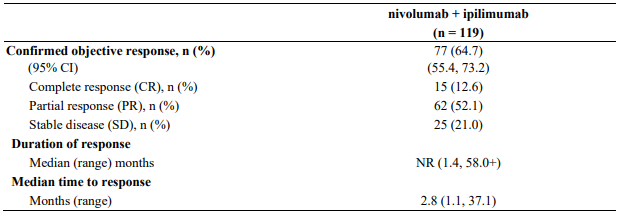
* per investigator assessment
"+" denotes a censored observation.
NR = not reached
The BICR-assessed ORR was 61.3% (95% CI: 52.0, 70.1), including CR rate of 20.2% (95% CI: 13.4, 28.5), PR rate of 41.2% (95% CI: 32.2, 50.6) and stable disease reported in 22.7%. BICR assessments were generally consistent with the investigator assessment. Confirmed responses were observed regardless of BRAF or KRAS mutation status, and tumour PD-L1 expression levels.
Of 119 patients 11 (9.2%) patients were ≥75 years. The investigator assessed ORR in patients ≥75 years was 45.5% (95% CI: 16.7, 76.6).
Oesophageal squamous cell carcinoma
Intravenous formulation
Randomised phase 3 study of nivolumab in combination with ipilimumab vs. chemotherapy and nivolumab in combination with chemotherapy vs. chemotherapy as first-line treatment (CA209648)
The safety and efficacy of nivolumab in combination with ipilimumab and nivolumab in combination with chemotherapy were evaluated in a randomised, active-controlled, open-label study (CA209648). The study included adult patients (18 years or older) with previously untreated, unresectable advanced, recurrent or metastatic OSCC. Patients were enrolled regardless of their tumour PD-L1 status, and tumour cell PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay. Patients were required to have squamous cell carcinoma or adenosquamous cell carcinoma of oesophagus, not amenable to chemoradiation and/or surgery. Prior adjuvant, neoadjuvant, or definitive chemotherapy, radiotherapy or chemoradiotherapy was permitted if given as part of curative intent regimen prior to trial enrollment. Patients who had a baseline performance score ≥2, had brain metastases that were symptomatic, had active autoimmune disease, used systemic corticosteroids or immunosuppressants, or patients at high risk of bleeding or fistula due to apparent invasion of tumour to organs adjacent to the oesophageal tumour were excluded from the study. Randomisation was stratified by tumour cell PD-L1 status (≥1% vs. <1% or indeterminate), region (East Asia vs. rest of Asia vs. rest of world), ECOG performance status (0 vs. 1), and number of organs with metastases (≤1 vs. ≥2).
A total of 970 patients were randomised to receive either nivolumab in combination with ipilimumab, (n=325), nivolumab in combination with chemotherapy (n=321), or chemotherapy (n=324). Of these, 473 patients had tumour cell PD-L1 expression ≥1%,158 in the nivolumab plus ipilimumab arm, 158 in the nivolumab plus chemotherapy arm, and 157 in the chemotherapy arm. Patients in the nivolumab plus ipilimumab arm received nivolumab 3 mg/kg every 2 weeks in combination with ipilimumab 1 mg/kg every 6 weeks, and patients in the nivolumab plus chemotherapy arm received nivolumab 240 mg every 2 weeks on days 1 and 15, fluorouracil 800 mg/m²/day intravenously on days 1 through 5 (for 5 days), and cisplatin 80 mg/m² intravenously on day 1 (of a 4-week cycle). Patients in the chemotherapy arm received fluorouracil 800 mg/m²/day intravenously on days 1 through 5 (for 5 days), and cisplatin 80 mg/m² intravenously on day 1 (of a 4-week cycle). Treatment continued until disease progression, unacceptable toxicity, or up to 24 months. Patients in the nivolumab plus ipilimumab arm who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue nivolumab as a single agent. Patients in the nivolumab plus chemotherapy arm in whom either fluorouracil and/or cisplatin were discontinued, other components of the treatment regimen were allowed to be continued.
Baseline characteristics were generally balanced across treatment groups. In patients with tumour cell PD-L1 expression ≥1%, the median age was 63 years (range: 26-85), 8.2% were ≥75 years of age, 81.8% were male, 73.1% were Asian, and 23.3% were white. Patients had histological confirmation of squamous cell carcinoma (98.9%) or adenosquamous cell carcinoma (1.1%) in the oesophagus. Baseline ECOG performance status was 0 (45.2%) or 1 (54.8%).
Nivolumab in combination with ipilimumab vs. chemotherapy
The primary efficacy outcome measures were PFS (by BICR) and OS assessed in patients with tumour cell PD-L1 expression ≥1%. Secondary endpoints per the pre-specified hierarchical testing included OS, PFS (by BICR), and ORR (by BICR) in all randomised patients. The tumour assessments per RECIST v1.1 were conducted every 6 weeks up to and including week 48, then every 12 weeks thereafter.
At the primary pre-specified analysis, with a minimum follow-up of 13.1 months, the study demonstrated a statistically significant improvement in OS in patients with tumour cell PD-L1 expression ≥1%. Efficacy results are shown in Table 37.
Table 37. Efficacy results in patients with tumour cell PD-L1 ≥1% (CA209648):
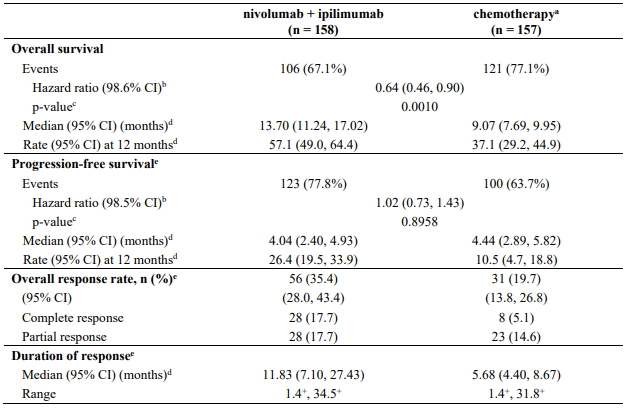
At an updated descriptive analysis with a minimum follow-up of 20 months, OS improvements were consistent with the primary analysis. Median OS was 13.70 months (95% CI: 11.24, 17.41) for nivolumab plus ipilimumab vs. 9.07 months (95% CI: 7.69, 10.02) for chemotherapy (HR = 0.63; 95% CI: 0.49, 0.82). Median PFS was 4.04 months (95% CI: 2.40, 4.93) for nivolumab plus ipilimumab vs. 4.44 months (95% CI: 2.89, 5.82) for chemotherapy (HR = 1.02; 95% CI: 0.77, 1.34). The ORR was 35.4% (95% CI: 28.0, 43.4) for nivolumab plus ipilimumab vs. 19.7% (95% CI: 13.8, 26.8) for chemotherapy.
The Kaplan-Meier curves for OS and PFS with a minimum follow-up of 20 months are shown in Figures 24 and 25.
Figure 24. Kaplan-Meier curves of OS in patients with tumour cell PD-L1 ≥1% (CA209648):
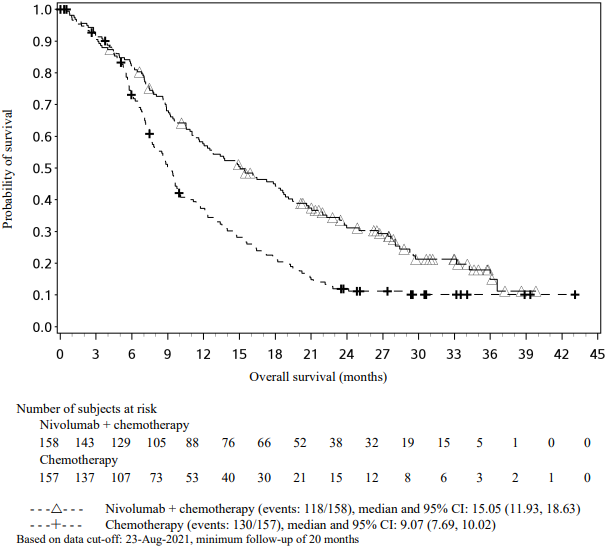
Figure 25. Kaplan-Meier curves of PFS in patients with tumour cell PD-L1 ≥1% (CA209648):
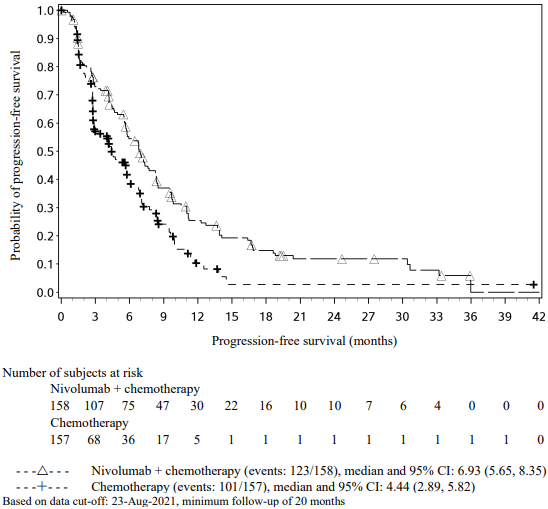
Intravenous formulation
Randomised phase 3 study of nivolumab monotherapy in previously treated patients (ONO-4538-24/CA209473)
The safety and efficacy of nivolumab 240 mg monotherapy for the treatment of unresectable advanced, recurrent or metastatic oesophageal squamous cell carcinoma (OSCC) was evaluated in a phase 3 randomised active-controlled, open-label study (ONO-4538-24/CA209473). The study included adult patients (20 years or older) who were refractory or intolerant to at least one fluoropyrimidine- and platinum-based combination regimen, and patients were enrolled regardless of tumour PD-L1 expression level. Patients who were refractory or intolerant to taxane therapy, had brain metastases that were symptomatic or required treatment, had active autoimmune disease, medical conditions requiring systemic immunosuppression, and patients with apparent tumour invasion in organs located adjacent to the oesophagus (e.g. the aorta or respiratory tract), were excluded from the study.
A total of 419 patients were randomised 1:1 to receive either nivolumab 240 mg administered intravenously over 30 minutes every 2 weeks (n=210) or investigator's choice of taxane chemotherapy: either docetaxel (n=65) 75 mg/m² intravenously every 3 weeks, or paclitaxel (n=144) 100 mg/m² intravenously once a week for 6 weeks followed by 1 week off. Randomisation was stratified by location (Japan vs. rest of world), number of organs with metastases (≤1 vs. ≥2) and tumour PD-L1 expression (≥1% vs. <1% or indeterminate). Treatment continued until disease progression, assessed by the investigator per RECIST version 1.1, or unacceptable toxicity. Tumour assessments were conducted every 6 weeks for 1 year, and every 12 weeks thereafter. Treatment beyond initial investigator-assessed progression was permitted in patients receiving nivolumab with no rapid progression, investigator-assessed benefit, tolerance to treatment, stable performance status, and for whom treatment beyond progression would not delay an imminent intervention to prevent serious complications associated with disease progression (e.g. brain metastasis). The primary efficacy outcome measure was OS. Key secondary efficacy outcome measures were investigator-assessed ORR and PFS. Additional prespecified subgroup analyses were conducted to evaluate the efficacy by tumour PD-L1 expression at a predefined level of 1%. Tumour PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay.
Baseline characteristics were generally balanced between the two groups. The median age was 65 years (range: 33-87), 53% were ≥65 years of age, 10% were aged ≥75 years, 87% were male, 96% were Asian and 4% were white. Baseline ECOG performance status was 0 (50%) or 1 (50%).
With a minimum follow-up of 17.6 months, the study demonstrated a statistically significant improvement in OS for patients randomised to nivolumab as compared with investigator's choice taxane chemotherapy. Efficacy results are shown in Table 38 and Figure 26.
A higher proportion of patients experienced death within the first 2.5 months in the nivolumab arm (32/210, 15.2%) as compared to the chemotherapy arm (15/209, 7.2%). No specific factor(s) associated with early deaths could be identified.
Table 38. Efficacy results (ONO-4538-24/CA209473):
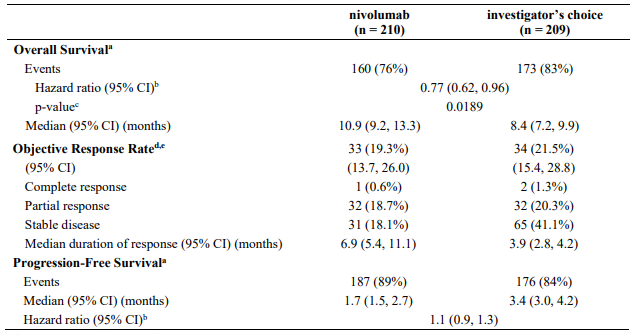
a Based on ITT analysis.
b Based on a stratified proportional hazards model.
c Based on a stratified log-rank test.
d Based on Response Evaluable Set (RES) analysis, n=171 in nivolumab group and n=158 in investigator's choice group.
e Not significant, p-value 0.6323.
Figure 26. Kaplan-Meier curves of OS (ONO-4538-24/CA209473):
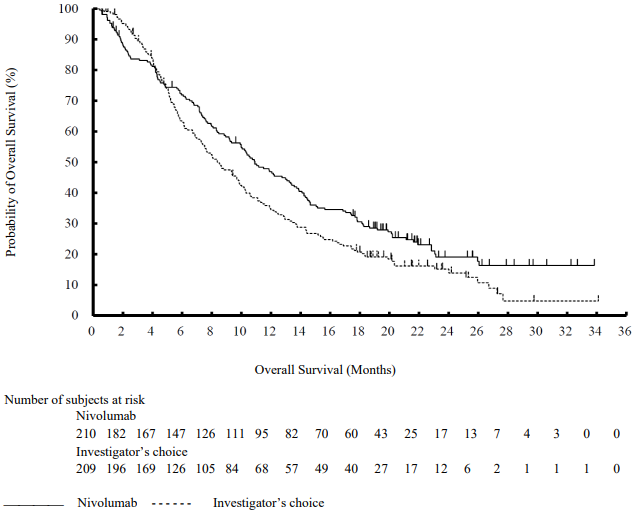
Of the 419 patients, 48% had tumour PD-L1 expression ≥1%. The remaining 52% of patients had tumour PD-L1 expression <1%. The hazard ratio (HR) for OS was 0.69 (95% CI: 0.51, 0.94) with median survivals of 10.9 and 8.1 months for the nivolumab and investigator's choice taxane chemotherapy arms, respectively, in the tumour PD-L1 positive subgroup. In the tumour PD-L1 negative OSCC subgroup, the HR for OS was 0.84 (95% CI: 0.62, 1.14) with median survivals of 10.9 and 9.3 months for the nivolumab and chemotherapy arms, respectively.
Adjuvant treatment of oesophageal or gastro-oesophageal junction cancer
Intravenous formulation
The safety and efficacy of nivolumab monotherapy for the adjuvant treatment of oesophageal or gastro-oesophageal junction cancer was evaluated in a phase 3 multicentre, randomised, placebo-controlled, double-blinded study (CA209577). The study included adult patients who had received CRT, followed by complete surgical resection of carcinoma within 16 weeks prior to randomisation, and who had residual pathologic disease as confirmed by the investigator, with at least ypN1 or ypT1. Patients with a baseline performance score ≥2, who did not receive concurrent CRT prior to surgery, with stage IV resectable disease, active autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. Patients were enrolled regardless of tumour PD-L1 expression level.
A total of 794 patients were randomised 2:1 to receive either nivolumab 240 mg (n=532) or placebo (n=262). Patients were administered nivolumab intravenously over 30 minutes every 2 weeks for 16 weeks followed by 480 mg infused over 30 minutes every 4 weeks beginning at week 17. Patients were administered placebo over 30 minutes with the same dosing schedule as nivolumab. Randomisation was stratified by tumour PD-L1 status (≥1% vs. <1% or indeterminate or non-evaluable), pathologic lymph node status (positive ≥ ypN1 vs. negative ypN0), and histology (squamous vs. adenocarcinoma). Treatment continued until disease recurrence, unacceptable toxicity, or for up to 1 year in total duration. The primary efficacy outcome measure was disease-free survival (DFS), as assessed by the investigator, defined as the time between the date of randomisation and the date of first recurrence (local, regional, or distant from the primary resected site) or death from any cause, whichever occurred first. Patients on treatment underwent imaging for tumour recurrence every 12 weeks for 2 years, and a minimum of one scan every 6 to 12 months for years 3 to 5.
Baseline characteristics were generally balanced between the two groups. The median age was 62 years (range: 26-86) with 36% ≥65 years of age and 5% ≥75 years of years. The majority of patients were white (82%) and male (85 %). Baseline ECOG performance status was 0 (58%) or 1 (42%).
At the primary pre-specified interim analysis (minimum of 6.2 months and a median of 24.4 months follow-up), the study demonstrated a statistically significant improvement in DFS for patients randomised to nivolumab compared with placebo. Median DFS as determined by the investigator was 22.41 months (95% CI: 16.62, 34.00) for nivolumab versus 11.04 months (95% CI: 8.34, 14.32) for placebo, HR 0.69 (96.4% CI: 0.56, 0.86), p-value <0.0003. The primary analysis of DFS included censoring for new anti-cancer treatment. Results for DFS with and without censoring for new anti-cancer treatment were consistent. In an updated descriptive DFS analysis with minimum of 14 months and median of 32.2 months follow-up, DFS improvement was confirmed. Efficacy results from this descriptive secondary analysis are shown in Table 39 and Figure 27.
Table 39. Efficacy results (CA209577):
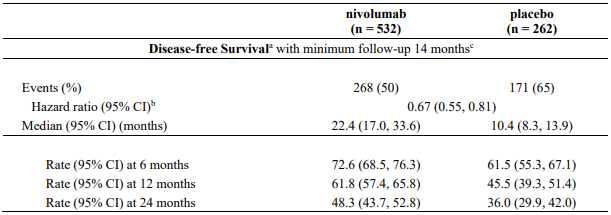
a Based on all randomised patients.
b Based on a stratified cox proportional hazards model.
c Descriptive analysis based on data cut-off: 18-Feb-2021.
Figure 27. Kaplan-Meier curves of DFS (CA209577):
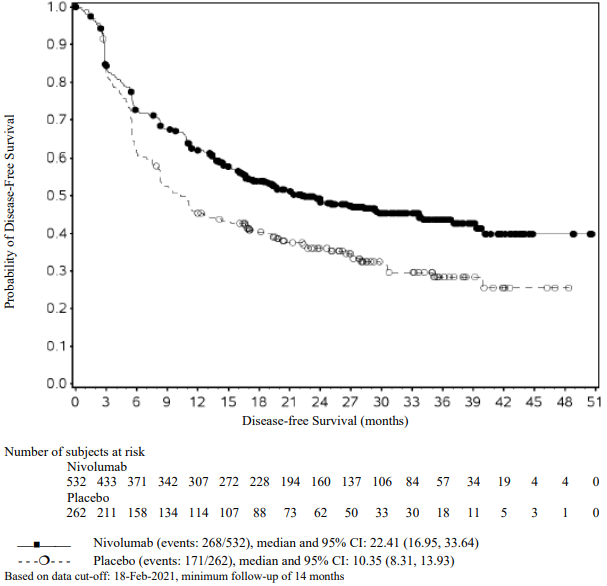
At the final OS analysis with a minimum follow up of 60 months, the HR for OS was 0.85 (95.87% CI: 0.70, 1.04), p-value = 0.1064. Median OS was 51.71 (95% CI: 41.03, 61.63) months in the nivolumab arm compared with 35.25 (95% CI: 30.72, 48.76) months in the placebo arm. The Kaplan-Meier curves for OS with a minimum follow-up of 60 months are shown in Figure 28.
Figure 28. Kaplan-Meier curves of OS (CA209577):
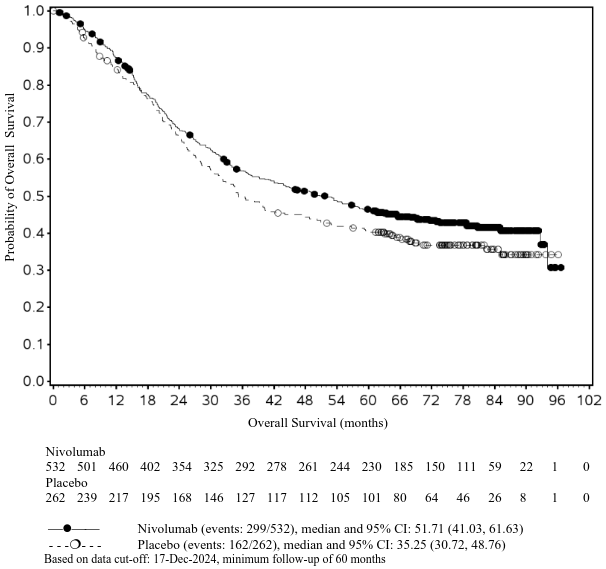
Gastric, gastro-oesophageal junction or oesophageal adenocarcinoma
Intravenous formulation
The safety and efficacy of nivolumab 240 mg every 2 weeks or 360 mg every 3 weeks in combination with chemotherapy (dose and schedule of nivolumab selected depending on the chemotherapy regimen used, see below) was evaluated in a phase 3, randomised, open-label study (CA209649). The study included adult patients (18 years or older) with previously untreated advanced or metastatic gastric, gastro-oesophageal junction (GEJ) or oesophageal adenocarcinoma, no prior systemic treatment (including HER2 inhibitors), and ECOG performance status score 0 or 1. Patients were enrolled regardless of their tumour cell PD-L1 status, and tumour cell PD-L1 expression was determined using the PD-L1 IHC 28-8 pharmDx assay. A retrospective re-scoring of a patient's tumour PD-L1 status using CPS was conducted using the PD-L1-stained tumour specimens used for randomisation. Patients with known HER2-positive tumours, who had baseline ECOG performance score ≥2, untreated central nervous system metastases, or who had active, known, or suspected autoimmune disease, or medical conditions requiring systemic immunosuppression were excluded from the study. A total of 643 patients with HER2-undetermined status (40.3% of the study population) were included in the study. Randomisation was stratified by tumour cell PD-L1 status (≥1% vs. <1% or indeterminate), region (Asia vs. US vs. rest of world), ECOG performance status (0 vs. 1), and chemotherapy regimen. Chemotherapy consisted of FOLFOX (fluorouracil, leucovorin and oxaliplatin) or CapeOX (capecitabine and oxaliplatin).
A total of 1581 patients were randomised to receive either nivolumab in combination with chemotherapy or chemotherapy. Of these, 955 patients had PD-L1 CPS ≥5; 473 in the nivolumab plus chemotherapy arm and 482 in the chemotherapy arm. Patients in the nivolumab plus chemotherapy arm received either nivolumab 240 mg by intravenous infusion over 30 minutes in combination with FOLFOX (oxaliplatin 85 mg/m², leucovorin 400 mg/m² and fluorouracil 400 mg/m² intravenously on day 1 and fluorouracil 1200 mg/m² intravenously by continuous infusion over 24 hours daily or per local standard on days 1 and 2) every 2 weeks, or nivolumab 360 mg by intravenous infusion over 30 minutes in combination with CapeOX (oxaliplatin 130 mg/m² intravenously on day 1 and capecitabine 1000 mg/m² orally twice daily on days 1-14) every 3 weeks. Treatment continued until disease progression, unacceptable toxicity, or for up to 24 months for nivolumab only. In patients who received nivolumab plus chemotherapy and in whom chemotherapy was discontinued, nivolumab monotherapy was allowed to be given at 240 mg every 2 weeks, 360 mg every 3 weeks or 480 mg every 4 weeks up to 24 months after treatment initiation. Tumour assessments were performed every 6 weeks up to and including week 48, then every 12 weeks thereafter.
Baseline characteristics were generally balanced across treatment groups. In patients with PD-L1 CPS ≥5, the median age was 62 years (range: 18-90), 11% were ≥75 years of age, 71% were male, 25% were Asian and 69% were white. Baseline ECOG performance status was 0 (42%) or 1 (58%). Tumour locations were distributed as gastric (70%), GEJ (18%) and oesophagus (12%).
Primary efficacy outcome measures were PFS (by BICR) and OS assessed in patients with PD-L1 CPS ≥5 based on the PD-L1 IHC 28-8 pharmDX. Secondary endpoints per the pre-specified hierarchical testing were OS in patients with PD-L1 CPS ≥1 and in all randomised patients; further endpoints included ORR (BICR) in PD-L1 CPS ≥5 and all randomised patients. At the primary prespecified analysis, with a minimum follow-up of 12.1 months, the study demonstrated a statistically significant improvement in OS and PFS in patients with PD-L1 CPS ≥5. Median OS was 14.4 months (95% CI: 13.1, 16.2) for nivolumab in combination with chemotherapy vs. 11.1 months (95% CI: 10.0, 12.1) for chemotherapy (HR = 0.71; 98.4% CI: 0.59, 0.86; p-value <0.0001). Median PFS was 7.69 months (95% CI: 7.03, 9.17) for nivolumab in combination with chemotherapy vs. 6.05 months (95% CI: 5.55, 6.90) for chemotherapy (HR = 0.68; 98% CI: 0.56, 0.81; p-value <0.0001). The ORR was 60% (95% CI: 55, 65) for nivolumab in combination with chemotherapy vs. 45% (95% CI: 40, 50) for chemotherapy.
At an updated descriptive analysis with a minimum follow-up of 19.4 months, OS improvements were consistent with the primary analysis. Efficacy results are shown in Table 40, and Figures 29 and 30.
Table 40. Efficacy results in patients with PD-L1 CPS ≥5 (CA209649):
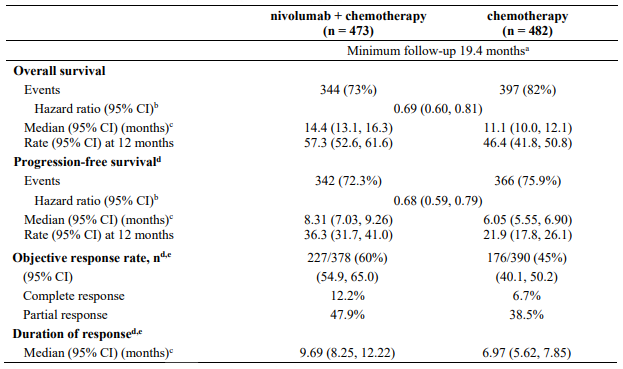
a Descriptive analysis based on data cut-off: 04-Jan-2021.
b Based on stratified long Cox proportional hazard model.
c Kaplan-Meier estimate.
d Confirmed by BICR. e Based on patients with measurable disease at baseline.
Figure 29. Kaplan-Meier curves of OS in patients with PD-L1 CPS ≥5 (CA209649):
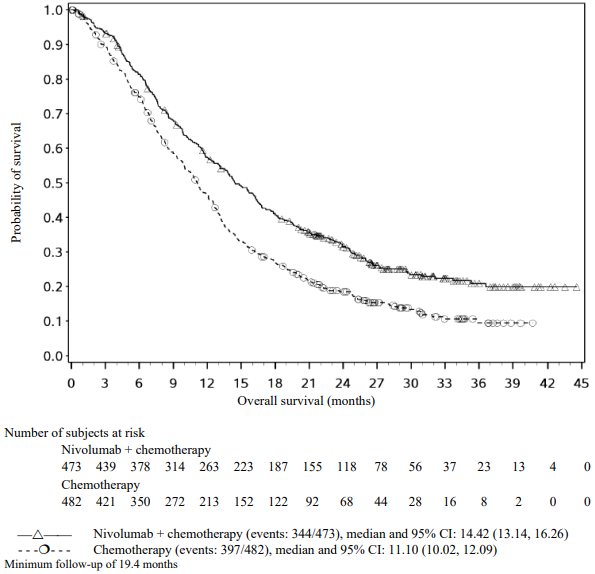
Figure 30. Kaplan-Meier curves of PFS in patients with PD-L1 CPS ≥5 (CA209649):
Hepatocellular carcinoma
Intravenous formulation
The safety and efficacy of nivolumab 1 mg/kg in combination with ipilimumab 3 mg/kg every 3 weeks, for a maximum of 4 doses, followed by nivolumab monotherapy 480 mg every 4 weeks in the first-line treatment of unresectable or advanced hepatocellular carcinoma (HCC) were evaluated in a phase 3, randomised, active-controlled, open-label study (CA2099DW). The study included adult patients (18 years or older) with histologically confirmed HCC, Child Pugh Class A, ECOG performance status 0 or 1, and no prior systemic therapy for advanced disease.
Esophagogastroduodenoscopy was not mandated prior to enrolment. The study enrolled adults whose disease was not amenable to or progressed after surgical and/or locoregional therapies. Prior neo-adjuvant or adjuvant systemic therapy was permitted. Patients with active autoimmune disease, brain or leptomeningeal metastases, prior liver transplant, a history of hepatic encephalopathy (within 12 months of randomisation), clinically significant ascites, medical conditions requiring systemic immunosuppression, infection with HIV, or active co infection with hepatitis B virus (HBV) and hepatitis C virus (HCV) or HBV and hepatitis D virus (HDV) were excluded from the study. Randomisation was stratified by aetiology (HBV vs. HCV vs. non-viral), macrovascular invasion and/or extrahepatic spread (present or absent), and alpha-fetoprotein levels (≥400 or <400 ng/mL).
A total of 668 patients were randomised to receive nivolumab in combination with ipilimumab (n=335) or investigator's choice (n=333) of lenvatinib or sorafenib. In the investigator's choice arm, 85% and 15% of treated patients received lenvatinib or sorafenib, respectively. Patients in the nivolumab plus ipilimumab arm received nivolumab 1 mg/kg every 3 weeks in combination with ipilimumab 3 mg/kg every 3 weeks, for up to a maximum of 4 doses, followed by nivolumab monotherapy 480 mg every 4 weeks. Patients in the investigators' choice arm received either lenvatinib 8 mg orally daily (if body weight <60 kg) or 12 mg orally daily (if body weight ≥60 kg), or sorafenib 400 mg orally twice daily. Treatment continued until disease progression, unacceptable toxicity, or for up to 24 months. Patients who discontinued combination therapy because of an adverse reaction attributed to ipilimumab were permitted to continue nivolumab as a single agent. Tumour assessments were conducted at baseline, after randomisation at week 9 and week 16, then every 8 weeks up to 48 weeks, and then every 12 weeks thereafter until disease progression, treatment discontinuation, or initiation of subsequent therapy.
Baseline characteristics were generally balanced across treatment groups. The median age was 66 years (range: 20 to 89), with 53% ≥65 years and 16% ≥75 years, 53% were White, 44% were Asian, 2.2% were Black, and 82% were male. Baseline ECOG performance status was 0 (71%) or 1 (29%). Thirty-four percent (34%) of patients had HBV infection, 28% had HCV infection, and 36% had no evidence of HBV or HCV infection. Nineteen percent (19%) of patients had alcoholic liver disease and 11% had non-alcoholic fatty liver disease. The majority of patients had BCLC stage C (73%) disease at baseline, 19% had stage B, and 6% had stage A. Patients with Child-Pugh scores of 5, 6, and ≥7 were 77%, 20%, and 3%, respectively. A total of 54% of patients had extrahepatic spread; 25% had macrovascular invasion; and 33% had AFP levels ≥400 μg/L.
The study demonstrated a statistically significant benefit in OS and ORR for patients randomised to nivolumab in combination with ipilimumab compared to investigator's choice of lenvatinib or sorafenib. Efficacy results are presented in Table 41 and Figure 31.
Table 41. Efficacy results in first-line HCC (CA2099DW)a:
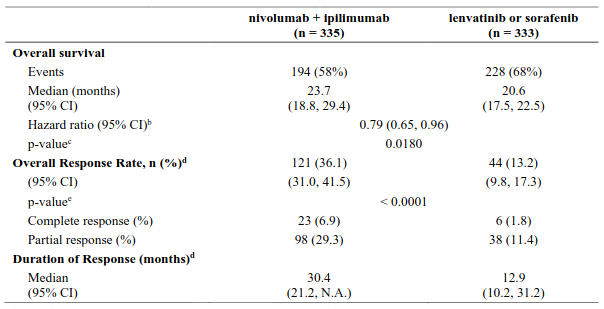
a Minimum follow-up of 26.8 months. Median follow up of 35.2 months.
b Based on stratified Cox proportional hazard model.
c Based on a 2-sided stratified log-rank test. Boundary for statistical significance: p-value ≤ 0.0257.
d Assessed by BICR using RECIST 1.1.
e Based on a 2-sided stratified Cochran-Mantel-Haenszel test. Boundary for statistical significance: p-value ≤ 0.025.
Figure 31. Kaplan-Meier curve of OS in first-line patients with HCC (CA2099DW):
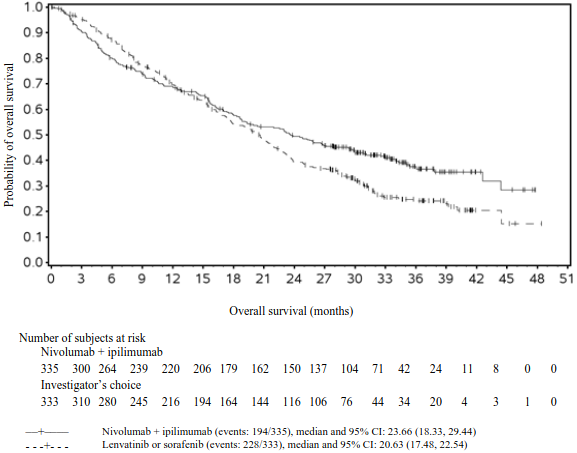
Paediatric population
Subcutaneous formulation
No dedicated studies of OPDIVO solution for injection have been conducted in paediatric patients.
The European Medicines Agency has waived the obligation to submit the results of studies with OPDIVO solution for injection for subcutaneous use in all subsets of the paediatric population in the treatment of malignant neoplasms (except central nervous system neoplasms, haematopoietic and lymphoid tissue neoplasms other than Hodgkin lymphoma) (see section 4.2 for information on paediatric use).
Safety and efficacy in elderly patients
No overall differences in safety or efficacy were reported between elderly (≥65 years) and younger patients (<65 years). Data from SCCHN, adjuvant melanoma, and adjuvant OC or GEJC patients 75 years of age or older are too limited to draw conclusions on this population.
5.2. Pharmacokinetic properties
Nivolumab solution for injection pharmacokinetics (PK) were assessed using a population PK approach. The PK was studied at a dose of 1200 mg administered as multiple doses every 4 weeks.
Nivolumab time-averaged serum concentration over 28 days (Cavgd28) showed non-inferiority of subcutaneous nivolumab (77.4 mcg/mL) to intravenous nivolumab (36.9 mcg/mL), with a geometric mean ratio of 2.098 (90% CI: 2.001, 2.200). Nivolumab minimum serum concentration at steady state (Cminss) showed non-inferiority of subcutaneous nivolumab (122.2 mcg/mL) to intravenous nivolumab (68.9 mcg/mL), with a geometric mean ratio of 1.774 (90% CI: 1.633, 1.927).
Absorption
The mean absorption rate constant (Ka) and bioavailability (F) of nivolumab solution for injection are 0.0123 hr-1 (or 0.295 Day-1) and 78.8%, respectively. Peak concentrations occurred by around 6 days.
Distribution
The geometric mean (CV%) volume of distribution at steady state (Vss) is 6.32 L (21.3%).
Elimination
Nivolumab solution for injection human clearance (CL) decreases over time, with a mean maximal reduction from baseline values (CV%) of 24.6% (15.8%) resulting in a geometric mean (CV%) steady-state clearance (CLss) of 7.18 mL/h (52.3%) in patients with RCC; the decrease in CLss is not considered clinically relevant.
The geometric mean (CV%) elimination half-life (t1/2) is 26.5 days (32.1%).
Special populations
The following factors had no clinically important effect on the bioavailability of nivolumab solution for injection: sex and performance status. The following factors had no clinically important effect on the clearance of nivolumab solution for injection: body weight (35 to 153 kg), sex, eGFR (24 to 124 mL/min/1.73 m²), or performance status.
Renal impairment
In population PK analyses for intravenous nivolumab, the effect of renal impairment on the CL of nivolumab was evaluated in patients with mild (GFR <90 and ≥60 mL/min/1.73 m²; n=379), moderate (GFR <60 and ≥30 mL/min/1.73 m²; n=179), or severe (GFR <30 and ≥15 mL/min/1.73 m²; n=2) renal impairment compared to patients with normal renal function (GFR ≥90 mL/min/1.73 m²; n=342). No clinically important differences in the CL of nivolumab were found between patients with mild or moderate renal impairment and patients with normal renal function. Data from patients with severe renal impairment are too limited to draw conclusions on this population (see section 4.2).
Hepatic impairment
In population PK analyses for intravenous nivolumab, the effect of hepatic impairment on the CL of nivolumab was evaluated in patients with different tumour types (NSCLC, SCLC, melanoma, RCC, SCCHN, UC, GC, and cHL) with mild hepatic impairment (total bilirubin 1.0 × to 1.5 × ULN or AST > ULN as defined using the National Cancer Institute criteria of hepatic dysfunction; n=351) and in patients with moderate hepatic impairment (total bilirubin >1.5 × to 3 × ULN and any AST; n=10) compared to patients with normal hepatic function (total bilirubin and AST ≤ ULN; n=3096). No clinically important differences in the CL of nivolumab were found between patients with mild or moderate hepatic impairment and normal hepatic function. Similar results were observed in patients with HCC (mild hepatic impairment: n=152; moderate hepatic impairment: n=13). Nivolumab has not been studied in patients with severe hepatic impairment (total bilirubin >3 × ULN and any AST) (see section 4.2).
5.3. Preclinical safety data
Blockade of PD-L1 signalling has been shown in murine models of pregnancy to disrupt tolerance to the foetus and to increase foetal loss. The effects of nivolumab on prenatal and postnatal development were evaluated in monkeys that received nivolumab twice weekly from the onset of organogenesis in the first trimester through delivery, at exposure levels either 8 or 35 times higher than those observed at the clinical dose of 3 mg/kg of nivolumab (based on AUC). There was a dose-dependent increase in foetal losses and increased neonatal mortality beginning in the third trimester.
The remaining offspring of nivolumab-treated females survived to scheduled termination, with no treatment-related clinical signs, alterations to normal development, organ-weight effects, or gross and microscopic pathology changes. Results for growth indices, as well as teratogenic, neurobehavioral, immunological, and clinical pathology parameters throughout the 6-month postnatal period were comparable to the control group. However, based on its mechanism of action, foetal exposure to nivolumab may increase the risk of developing immune-related disorders or altering the normal immune response and immune-related disorders have been reported in PD-1 knockout mice.
Fertility studies have not been performed with nivolumab.
Subcutaneous formulation
Hyaluronidase is found in most tissues of the human body. Non-clinical data for recombinant human hyaluronidase reveal no special hazard for humans based on conventional studies of repeated dose toxicity including safety pharmacology endpoints. Reproductive toxicology studies with rHuPH20 revealed embryofoetal toxicity in mice at high systemic exposure but did not show teratogenic potential.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.