ORILISSA Film-coated tablet Ref.[10902] Active ingredients: Elagolix
Source: FDA, National Drug Code (US) Revision Year: 2021
12.1. Mechanism of Action
ORILISSA is a GnRH receptor antagonist that inhibits endogenous GnRH signaling by binding competitively to GnRH receptors in the pituitary gland. Administration of ORILISSA results in dose-dependent suppression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH), leading to decreased blood concentrations of the ovarian sex hormones, estradiol and progesterone.
12.2. Pharmacodynamics
Effect on Ovulation and Estradiol
In a 3-menstrual cycle study in healthy women, ORILISSA 150 mg once daily and 200 mg twice daily resulted in an ovulation rate of approximately 50% and 32%, respectively. In the Phase 3 trials in women with endometriosis, ORILISSA caused a dose-dependent reduction in median estradiol concentrations to approximately 42 pg/mL for 150 mg once daily regimen and 12 pg/mL for the 200 mg twice daily regimen.
Cardiac Electrophysiology
The effect of elagolix on the QTc interval was evaluated in a randomized, placebo- and positive-controlled, open-label, single-dose, crossover thorough QTc study in 48 healthy adult premenopausal women. Elagolix concentrations in subjects given a single dose of 1200 mg was 17-times higher than the concentration in subjects given elagolix 200 mg twice daily. There was no clinically relevant prolongation of the QTc interval.
12.3. Pharmacokinetics
The pharmacokinetic properties of ORILISSA in healthy subjects are summarized in Table 8. The steady state pharmacokinetic parameters under fasting conditions are summarized in Table 9.
Table 8. Pharmacokinetic Properties of ORILISSA in Healthy Subjects:
| Absorption | |
|---|---|
| Tmax (h) | 1.0 |
| Effect of high-fat meal (relative to fasting) | AUC: ↓24%, Cmax: ↓36% |
| Distribution | |
| % Bound to human plasma proteins | 80 |
| Blood-to-plasma ratio | 0.6 |
| Metabolism | |
| Metabolism | CYP3A (major) Minor pathways include: CYP2D6, CYP2C8, and uridine glucuronosyl transferases (UGTs) |
| Elimination | |
| Major route of elimination | Hepatic metabolism |
| Terminal phase elimination half-life (t1/2) (h) | 4-6 |
| % of dose excreted in urine | <3 |
| % of dose excreted in feces | 90 |
Table 9. Mean (%CV) Steady State Pharmacokinetic Parameters of ORILISSA:
| Pharmacokinetic Parameter (Units) | 150 mg Once Daily N=6 | 200 mg Twice Daily N=7 |
|---|---|---|
| Cmax (ng/mL) | 574 (29) | 774 (68) |
| AUCτ (ng●hr/mL) | 1292 (31) | 1725 (57) |
| CL/F (L/hr) | 123 (21) | 144 (43) |
| Vdss/F | 1674 (94) | 881 (38) |
| Rac | 0.98 (7) | 0.89 (19) |
CV: Coefficient of variation
Cmax: peak concentration
AUCτ: area under the plasma concentration-time curve during the dosing interval (τ) i.e., 12 hours for twice daily regimen, 24 hours for once daily regimen.
CL/F: oral clearance
Vdss/F: apparent volume of distribution at steady state
Rac: drug accumulation ratio
Specific Populations
Patients with Renal Impairment
Elagolix exposures (Cmax and AUC) are not altered by renal impairment. The mean exposures are similar for women with moderate to severe or end stage renal disease (including women on dialysis) compared to women with normal renal function.
Patients with Hepatic Impairment
Elagolix exposures (Cmax and AUC) are similar between women with normal hepatic function and women with mild hepatic impairment. Elagolix exposures in women with moderate and severe hepatic impairment are approximately 3-fold and 7-fold, respectively, higher than exposures from women with normal hepatic function [see Use in Specific Populations (8.7)].
Racial or Ethnic Groups
No clinically meaningful difference in the pharmacokinetics of ORILISSA between White and Black subjects or between Hispanics and others was observed. There is no clinically meaningful difference in the pharmacokinetics of ORILISSA between Japanese and Han Chinese subjects.
Body weight/Body mass index
Body weight or body mass index does not affect the pharmacokinetics of ORILISSA.
Drug Interaction Studies
Drug interaction studies were performed with ORILISSA and other drugs that are likely to be co-administered and with drugs commonly used as probes for pharmacokinetic interactions. Tables 10 and 11 summarize the pharmacokinetic effects when elagolix was co-administered with these drugs.
Table 10. Drug Interactions: Change in Pharmacokinetics of Elagolix in the Presence of Co-administered Drugs:
| Co-administered Drug | Regimen of Co-administered Drug | Regimen of Elagolix | N | Ratio (90% CI)* | |
|---|---|---|---|---|---|
| Cmax | AUC | ||||
| Ketoconazole | 400 mg once daily | 150 mg single dose | 11 | 1.77 (1.48–2.12) | 2.20 (1.98–2.44) |
| Rifampin# | 600 mg single dose | 150 mg single dose | 12 | 4.37 (3.62–5.28) | 5.58 (4.88–6.37) |
| 600 mg once daily | 2.00 (1.66–2.41) | 1.65 (1.45–1.89) | |||
CI: Confidence interval
* ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of elagolix alone.
# A single dose of 600 mg rifampin inhibits OATP1B1; 600 mg once daily dose of rifampin inhibits OATP1B1 and induces CYP3A.
No clinically significant changes in elagolix exposures were observed when co-administered with rosuvastatin (20 mg once daily), sertraline (25 mg once daily) or fluconazole (200 mg single dose).
Table 11. Drug Interactions: Change in Pharmacokinetics of Co-administered Drug in the Presence of Elagolix:
| Co-administered Drug | Regimen of Co-administered Drug | Regimen of Elagolix | N | Ratio (90% CI)* | |
|---|---|---|---|---|---|
| Cmax | AUC | ||||
| Digoxin | 0.5 mg single dose | 200 mg twice daily x 10 days | 11 | 1.71 (1.53–1.91) | 1.26 (1.17–1.35) |
| Rosuvastatin | 20 mg once daily | 300 mg twice daily x 7 days | 10 | 0.99 (0.73–1.35) | 0.60 (0.50–0.71) |
| Midazolam | 2 mg single dose | 300 mg twice daily x 11 days | 20 | 0.56 (0.51–0.62) | 0.46 (0.41–0.50) |
| 150 mg once daily x 13 days | 11 | 0.81 (0.74–0.89) | 0.65 (0.58–0.72) | ||
| Norethindrone | 0.35 mg once daily x 112 days | 150 mg once daily x 56 days | 32 | 0.95 (0.86–1.05) | 0.88 (0.79–0.99) |
| Ethinyl Estradiol | Ethinyl estradiol 35 mcg and triphasic norgestimate 0.18/0.215/0.25 mg once daily | 150 mg once daily | 21 | 1.15 (1.07–1.25) | 1.30 (1.19–1.42) |
| Norelgestromina | 0.87 (0.78–0.97) | 0.85 (0.78–0.92) | |||
| Norgestrela | 0.89 (0.78–1.00) | 0.92 (0.84–1.01) | |||
| Ethinyl Estradiol | Ethinyl estradiol 20 mcg/Levonorgestrel 0.1 mg single dose | 200 mg twice daily x 15 days | 20 | 1.36 (1.27–1.45) | 2.18 (1.99–2.39) |
| Levonorgestrel | 0.97 (0.88–1.07) | 0.73 (0.64–0.82) | |||
| Omeprazole | 40 mg single dose | 300 mg twice daily x 9 days | 20 | 1.95 (1.50–2.53) | 1.78 (1.39–2.27) |
CI: Confidence interval
* ratios for Cmax and AUC compare co-administration of the medication with elagolix vs. administration of the medication alone.
a metabolite of norgestimate
No clinically significant changes were observed in exposures of sertraline, fluconazole, or bupropion when co-administered with elagolix 300 mg twice daily.
12.5. Pharmacogenomics
Hepatic uptake of elagolix involves the OATP 1B1 transporter protein. Higher plasma concentrations of elagolix have been observed in patients who have two reduced function alleles of the gene that encodes OATP 1B1 (SLCO1B1 521T>C) (these patients are likely to have reduced hepatic uptake of elagolix and thus, higher plasma elagolix concentrations). The frequency of this SLCO1B1 521 C/C genotype is generally less than 5% in most racial/ethnic groups. Subjects with this genotype are expected to have a 78% mean increase in elagolix concentrations compared to subjects with normal transporter function (i.e., SLCO1B1 521T/T genotype). Adverse effects of elagolix have not been fully evaluated in subjects who have two reduced function alleles of the gene that encodes OATP1B1 (SLCO1B1 521T>C).
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies conducted in mice (50, 150, or 500 mg/kg/day) and rats (150, 300, or 800 mg/kg/day) that administered elagolix by the dietary route revealed no increase in tumors in mice at up to 19-fold the MRHD based on AUC. In the rat, there was an increase in thyroid (male and female) and liver (males only) tumors at the high dose (12 to 13-fold the MRHD). The rat tumors were likely species-specific and of negligible relevance to humans.
Elagolix was not genotoxic or mutagenic in a battery of tests, including the in vitro bacterial reverse mutation assay, the in vitro mammalian cell forward mutation assay at the thymidine kinase (TK+/-) locus in L5178Y mouse lymphoma cells, and the in vivo mouse micronucleus assay.
In a fertility study conducted in the rat, there was no effect of elagolix on fertility at any dose (50, 150, or 300 mg/kg/day). Based on AUC, the exposure multiple for the MRHD in women compared to the highest dose of 300 mg/kg/day in female rats is approximately 5-fold. However, because elagolix has low affinity for the GnRH receptor in the rat [see Use in Specific Populations (8.1)], and because effects on fertility are most likely to be mediated via the GnRH receptor, these data have low relevance to humans.
14. Clinical Studies
The efficacy of ORILISSA 150 mg once daily and 200 mg twice daily for the management of moderate to severe pain associated with endometriosis was demonstrated in two multinational double-blind, placebo-controlled trials in 1686 premenopausal women [Study EM-1 (NCT01620528) and Study EM-2 (NCT01931670)]. The median age of women in the trials was 32 years; 88% were White, 9% were Black or African American and 3% were other races. Each placebo-controlled trial assessed the reduction in endometriosis-associated pain over 6 months of treatment.
Moderate to severe pain associated with endometriosis was required for entry into the trials and was assessed during screening using the composite pelvic signs and symptoms score (CPSSS) and other baseline criteria.
The CPSSS is based on a modified Biberoglu and Behrman scale with five elements: three responses reported by study subjects (dysmenorrhea, dyspareunia, and non-menstrual pelvic pain) and two findings based on investigator assessment during physical examination (rating of pelvic tenderness and induration). Each element is scored from 0 (absent) to 3 (severe) for a maximum total score of 15. A total score of at least 6, with a score of at least 2 for dysmenorrhea and at least 2 for non-menstrual pelvic pain was required to qualify for randomization. Subjects were also required to have non-menstrual pelvic pain for at least four days in the preceding calendar month, defined as 35 days. Other criteria to determine eligibility for randomization included subject responses in a daily electronic diary (Endometriosis Daily Pain Impact Scale, described below) for both dysmenorrhea and non-menstrual pelvic pain in the 35 days prior to randomization.
Dysmenorrhea and Non-Menstrual Pelvic Pain
The co-primary efficacy endpoints were (1) the proportion of subjects whose dysmenorrhea responded to treatment at Month 3 and (2) the proportion of subjects whose pelvic pain not related to menses (also known as non-menstrual pelvic pain) responded to treatment at Month 3. Dysmenorrhea and non-menstrual pelvic pain were evaluated daily using the Endometriosis Daily Pain Impact Scale that asked subjects to rate their pain severity and its impact on daily activities during the prior 24 hours as none, mild, moderate or severe (correlating with a score of 0 to 3, respectively, where higher scores indicated greater severity). Scores at baseline and at each month were averaged over a 35-day interval.
Women were defined as responders if they experienced a reduction in dysmenorrhea and non-menstrual pelvic pain as defined in Table 12 with no increase in analgesic use (nonsteroidal anti-inflammatory drug or opioid) for endometriosis-associated pain. The threshold for defining responders was based on a receiver operating characteristic (ROC) analysis using the patient global impression of change as an anchor. A higher proportion of women treated with ORILISSA 150 mg once daily or 200 mg twice daily were responders for dysmenorrhea and non-menstrual pelvic pain compared to placebo in a dose-dependent manner at Month 3 [see Table 12].
Table 12. Proportion of Responders† for Dysmenorrhea and Non-Menstrual Pelvic Pain at Month 3 in Studies EM-1 and EM-2, Using the Endometriosis Daily Pain Impact Scale:
| Study EM-1 | Study EM-2 | |||||
|---|---|---|---|---|---|---|
| ORILISSA | Placebo | ORILISSA | Placebo | |||
| 150 mg Once Daily N=248 | 200 mg Twice Daily N=244 | N=373 | 150 mg Once Daily N=221 | 200 mg Twice Daily N=225 | N=353 | |
| Dysmenorrhea Difference from placebo | 46% 27%** | 76% 56%** | 20% | 43% 21%** | 72% 50%** | 23% |
| Non-Menstrual Pelvic Pain Difference from placebo | 50% 14%** | 55% 18%** | 36% | 50% 13%* | 58% 21%** | 37% |
† Study EM-1-Dysmenorrhea responder threshold: at least 0.81 point decrease from baseline in dysmenorrhea score; Non-Menstrual Pelvic Pain responder threshold: at least 0.36 point decrease from baseline in Non-Menstrual Pelvic Pain score
Study EM-2 - Dysmenorrhea responder threshold: at least 0.85 point decrease from baseline in dysmenorrhea score; Non-Menstrual Pelvic Pain responder threshold: at least 0.43 point decrease from baseline in Non-Menstrual Pelvic Pain score
* p≤0.01 for test of difference from placebo
** p≤0.001 for test of difference from placebo
Women in these studies also provided a daily self-assessment of their endometriosis pain using a numeric rating scale (NRS) that asked subjects to rate their endometriosis pain at its worst over the last 24 hours on a scale from 0 (no pain) to 10 (worst pain ever). In Study EM-1, baseline NRS scores were 5.7 for ORILISSA 150 mg once daily, 5.5 for ORILISSA 200 mg twice daily and 5.6 for placebo. In Study EM-2, baseline NRS scores were 5.7 for ORILISSA 150 mg once daily, 5.3 for ORILISSA 200 mg twice daily and 5.6 for placebo. Women taking ORILISSA 150 mg once daily and 200 mg twice daily reported a statistically (p<0.001) significant reduction from baseline in NRS scores compared to placebo at Month 3 in both Studies EM-1 and EM-2 (Study EM-1: 0.7 points for ORILISSA 150 mg once daily and 1.3 points for ORILISSA 200 mg twice daily; Study EM-2: 0.6 points for ORILISSA 150 mg once daily and 1.2 points for ORILISSA 200 mg twice daily).
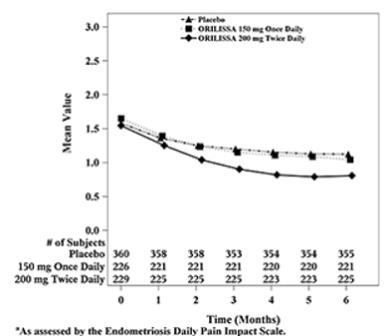
In addition, both ORILISSA treatment groups showed statistically significantly greater mean decreases from baseline compared to placebo in dysmenorrhea and non-menstrual pelvic pain scores at Month 6. Figures 3 through 6 show the mean scores for dysmenorrhea and non-menstrual pelvic pain over time for Study EM-1 and EM-2.
Figure 3. Mean Dysmenorrhea Pain Scoresa in Study EM-1 Over 6 Months:
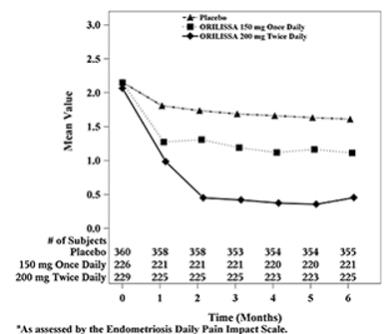
Figure 4. Mean Dysmenorrhea Pain Scoresa in Study EM-2 Over 6 Months:
Figure 5. Mean Non-Menstrual Pelvic Paina Scores in Study EM-1 Over 6 Months:
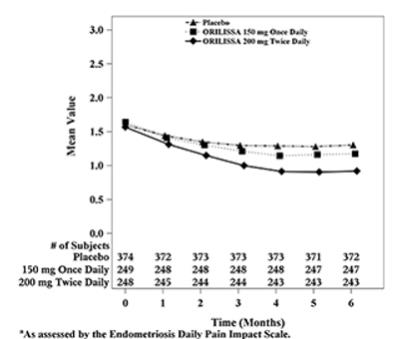
Figure 6. Mean Non-Menstrual Pelvic Paina Scores in Study EM-2 Over 6 Months:
Dyspareunia
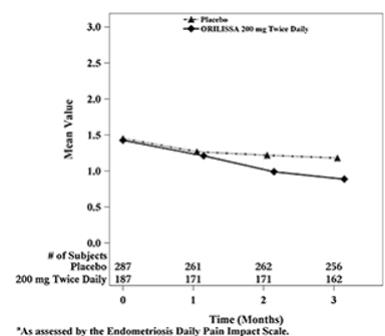
Dyspareunia associated with endometriosis was evaluated as a secondary endpoint using the Endometriosis Daily Pain Impact Scale that asked subjects to rate their pain during sexual intercourse in the prior 24 hours as none, mild, moderate, severe (correlating with a score of 0 to 3, respectively, where higher scores indicated greater severity), or not applicable. In both Studies EM-1 and EM-2, women treated with ORILISSA 200 mg twice daily showed statistically significantly greater reduction in dyspareunia from baseline to Month 3 than women given placebo (Study EM-1: 0.2; Study EM-2: 0.3). Figures 7 and 8 show the mean scores over time for Study EM-1 and EM-2.
Figure 7. Mean Dyspareunia Scoresa in Study EM-1 Over 3 Months:
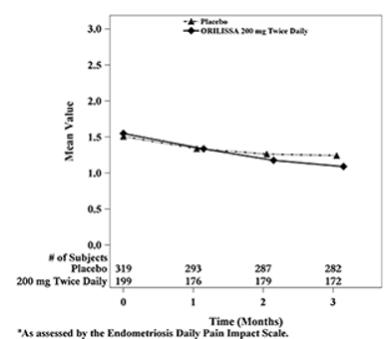
Figure 8. Mean Dyspareunia Scoresa in Study EM-2 Over 3 Months:
Use of rescue pain medication
In EM-1 and EM-2, 59% and 60% of patients used an opioid rescue analgesic for pain at baseline. The opioid rescue analgesics used at baseline were predominantly hydrocodone/acetaminophen (HC/APAP) and codeine/APAP at strengths of 5/300-325 mg and 30/300-500 mg. In EM-1, of all patients on an opioid at baseline, 98% and 2% were on HC/APAP and codeine/APAP, respectively. In EM-2, of all patients on an opioid at baseline, 50% were on HC/APAP and 16% were on codeine/APAP.
Other data related to opioid rescue analgesic use are summarized in Table 13.
Table 13. Opioid Rescue Analgesic Use in EM-1 and EM-2:
| Study EM-1 | Study EM-2 | |||||
|---|---|---|---|---|---|---|
| ORILISSA 150 mg Once Daily | ORILISSA 200 mg Twice Daily | Placebo | ORILISSA 150 mg Once Daily | ORILISSA 200 mg Twice Daily | Placebo | |
| Tablets per month at baseline (mean±SD) | 15 ±24 | 15 ±25 | 13 ±21 | 13 ±29 | 12 ±26 | 12 ±21 |
| Tablets per month at baseline [Median (Min, Max)] | 4 (0, 184) | 4 (0, 195) | 4 (0, 146) | 4 (0, 236) | 3 (0, 214) | 4 (0, 152) |
| Tablets per month at Month 3 (mean±SD) | 12 ±29 | 7 ±18 | 10 ±17 | 8 ±22 | 5 ±14 | 8 ±15 |
| Tablets per month at Month 3 [Median (Min, Max)] | 0 (0, 251) | 0 (0, 162) | 2 (0, 144) | 0 (0, 168) | 0 (0, 136) | 2 (0, 142) |
| Tablets per month at Month 6 (mean±SD) | 11 ±26 | 7 ±17 | 11 ±19 | 7 ±19 | 5 ±14 | 8 ±15 |
| Tablets per month at Month 6 [Median (Min, Max)] | 0 (0, 224) | 0 (0, 157) | 3 (0, 185) | 0 (0, 185) | 0 (0, 157) | 2 (0, 142) |
| Number and % of patients on any dose of opioid rescue at baseline who were off opioid at Month 3* | 46/150 (31%) | 59/151 (39%) | 36/211 (17%) | 44/124 (35%) | 68/134 (51%) | 54/220 (25%) |
| Number and % of patients on any dose of opioid rescue at baseline who were off opioid at Month 6* | 43/149 (29%) | 66/150 (44%) | 36/211 (17%) | 50/124 (40%) | 78/134 (58%) | 70/222 (32%) |
| Number and % of patients not on opioid rescue at baseline who were on any opioid at Month 3^ | 9/98 (9%) | 6/93 (6%) | 17/162 (10%) | 10/97 (10%) | 10/91 (11%) | 29/133 (22%) |
| Number and % of patients not on opioid rescue at baseline who were on any opioid at Month 6^ | 16/98 (16%) | 6/93 (6%) | 32/161 (20%) | 13/97 (13%) | 6/91 (7%) | 32/133 (24%) |
Min = minimum; Max = maximum; SD = standard deviation
Monthly calculations are based on a 35-day interval.
* Denominator is the number of subjects on opioid rescue at baseline.
^ Denominator is the number of subjects not on opioid rescue at baseline.
The clinical relevance of these data has not been demonstrated.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.