Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Vertex Pharmaceuticals (Ireland) Limited, Unit 49, Block F2, Northwood Court, Santry, Dublin 9, D09 T665, Ireland
Hypersensitivity to the active substances or to any of the excipients listed in section 6.1.
Lumacaftor/ivacaftor is not effective in patients with CF who have the F508del mutation on one allele plus a second allele with a mutation predicted to result in a lack of CFTR production or that is not responsive to ivacaftor in vitro (see section 5.1).
Lumacaftor/ivacaftor has not been studied in patients with CF who have a gating (Class III) mutation in the CFTR gene on one allele, with or without the F508del mutation on the other allele. Since the exposure of ivacaftor is very significantly reduced when dosed in combination with lumacaftor, lumacaftor/ivacaftor should not be used in these patients.
Respiratory adverse reactions (e.g., chest discomfort, dyspnoea, bronchospasm, and respiration abnormal) were more common during initiation of lumacaftor/ivacaftor therapy. Serious respiratory events were seen more frequently in patients with percent predicted forced expiratory volume in 1 second (ppFEV1) <40, and may lead to discontinuation of the medicinal product. Clinical experience in patients with ppFEV1 <40 is limited and additional monitoring of these patients is recommended during initiation of therapy (see section 4.8). A transient decline in FEV1 has also been observed in some patients following initiation of lumacaftor/ivacaftor. There is no experience of initiating treatment with lumacaftor/ivacaftor in patients having a pulmonary exacerbation and initiating treatment in patients having a pulmonary exacerbation is not advisable.
Increased blood pressure has been observed in some patients treated with lumacaftor/ivacaftor. Blood pressure should be monitored periodically in all patients during treatment (see section 4.8).
Abnormalities in liver function, including advanced liver disease, can be present in patients with CF. Worsening of liver function in patients with advanced liver disease has been reported. Liver function decompensation, including liver failure leading to death, has been reported in CF patients with preexisting cirrhosis with portal hypertension receiving lumacaftor/ivacaftor. Lumacaftor/ivacaftor should be used with caution in patients with advanced liver disease and only if the benefits are expected to outweigh the risks. If lumacaftor/ivacaftor is used in these patients, they should be closely monitored after the initiation of treatment and the dose should be reduced (see sections 4.2, 4.8, and 5.2).
Elevated transaminases have been commonly reported in patients with CF receiving lumacaftor/ivacaftor. In some instances, these elevations have been associated with concomitant elevations in total serum bilirubin. Transaminase elevations have been observed more frequently in paediatric patients than in adult patients (see section 4.8).
Because an association with liver injury cannot be excluded, assessments of liver function tests (ALT, AST and bilirubin) are recommended before initiating lumacaftor/ivacaftor, every 3 months during the first year of treatment, and annually thereafter. For patients with a history of ALT, AST, or bilirubin elevations, more frequent monitoring should be considered.
In the event of significant elevation of ALT or AST, with or without elevated bilirubin (either ALT or AST >5 x the upper limit of normal [ULN], or ALT or AST >3 x ULN with bilirubin >2 x ULN and/or clinical jaundice), dosing with lumacaftor/ivacaftor should be discontinued and laboratory tests closely followed until the abnormalities resolve. A thorough investigation of potential causes should be conducted and patients should be followed closely for clinical progression. Following resolution of transaminase elevations, the benefits and risks of resuming dosing should be considered (see sections 4.2, 4.8, and 5.2).
Lumacaftor is a strong inducer of CYP3A. Co-administration with sensitive CYP3A substrates or CYP3A substrates with a narrow therapeutic index is not recommended (see section 4.5). Hormonal contraceptives, including oral, injectable, transdermal, and implantable, should not be relied upon as an effective method of contraception when co-administered with Orkambi (see section 4.5).
Ivacaftor is a substrate of CYP3A4 and CYP3A5. Therefore, co-administration with strong CYP3A inducers (e.g., rifampicin, St. John’s wort [Hypericum perforatum]) is not recommended (see section 4.5).
Caution is recommended while using lumacaftor/ivacaftor in patients with severe renal impairment or end-stage renal disease (see sections 4.2 and 5.2).
Cases of non-congenital lens opacities without impact on vision have been reported in paediatric patients treated with lumacaftor/ivacaftor and ivacaftor monotherapy. Although other risk factors were present in some cases (such as corticosteroid use and exposure to radiation), a possible risk attributable to ivacaftor cannot be excluded (see section 5.3). Baseline and follow-up ophthalmological examinations are recommended in paediatric patients initiating treatment with lumacaftor/ivacaftor.
Lumacaftor/ivacaftor has not been studied in patients with CF who have undergone organ transplantation. Therefore, use in transplanted patients is not recommended. See section 4.5 for interactions with immunosuppressants.
This medicinal product contains less than 1 mmol sodium (23 mg) per dose, that is to say essentially ‘sodium-free’.
Based on exposure and indicated doses, the interaction profile is considered to be the same for all strengths and pharmaceutical forms.
Lumacaftor is a strong inducer of CYP3A and ivacaftor is a weak inhibitor of CYP3A when given as monotherapy. There is potential for other medicinal products to affect lumacaftor/ivacaftor when administered concomitantly, and also for lumacaftor/ivacaftor to affect other medicinal products.
Co-administration of lumacaftor/ivacaftor with itraconazole, a strong CYP3A inhibitor, did not impact the exposure of lumacaftor, but increased ivacaftor exposure by 4.3-fold. Due to the induction effect of lumacaftor on CYP3A, at steady-state, the net exposure of ivacaftor when co-administered with a CYP3A inhibitor is not expected to exceed that when given in the absence of lumacaftor at a dose of 150 mg every 12 hours, the approved dose of ivacaftor monotherapy.
No dose adjustment is necessary when CYP3A inhibitors are initiated in patients currently taking lumacaftor/ivacaftor. However, when initiating lumacaftor/ivacaftor in patients taking strong CYP3A inhibitors, the dose should be adjusted (see sections 4.2 and 4.4).
No dose adjustment is recommended when used with moderate or weak CYP3A inhibitors.
Co-administration of lumacaftor/ivacaftor with rifampicin, a strong CYP3A inducer, had minimal effect on the exposure of lumacaftor, but decreased ivacaftor exposure (AUC) by 57%. Therefore, co-administration of lumacaftor/ivacaftor is not recommended with strong CYP3A inducers (see sections 4.2 and 4.4). No dose adjustment is recommended when used with moderate or weak CYP3A inducers.
Lumacaftor is a strong inducer of CYP3A. Ivacaftor is a weak inhibitor of CYP3A when given as monotherapy. The net effect of lumacaftor/ivacaftor therapy is expected to be strong CYP3A induction. Therefore, concomitant use of lumacaftor/ivacaftor with CYP3A substrates may decrease the exposure of these substrates (see section 4.4).
In vitro studies indicated that lumacaftor has the potential to both inhibit and induce P-gp. Additionally, a clinical study with ivacaftor monotherapy showed that ivacaftor is a weak inhibitor of P-gp. Therefore, concomitant use of lumacaftor/ivacaftor with P-gp substrates (e.g., digoxin) may alter the exposure of these substrates.
Interaction with CYP2B6 and CYP2C substrates has not been investigated in vivo. In vitro studies suggest that lumacaftor has the potential to induce CYP2B6, CYP2C8, CYP2C9, and CYP2C19; however, inhibition of CYP2C8 and CYP2C9 has also been observed in vitro. Additionally, in vitro studies suggest that ivacaftor may inhibit CYP2C9. Therefore, concomitant use of lumacaftor/ivacaftor may alter (i.e., either increase or decrease) the exposure of CYP2C8 and CYP2C9 substrates, decrease the exposure of CYP2C19 substrates, and substantially decrease the exposure of CYP2B6 substrates.
In vitro experiments show that lumacaftor is a substrate for Breast Cancer Resistance Protein (BCRP). Co-administration of Orkambi with medicinal products that inhibit BCRP may increase plasma lumacaftor concentration. Lumacaftor inhibits the organic anion transporter (OAT) 1 and 3. Lumacaftor and ivacaftor are inhibitors of BCRP. Co-administration of Orkambi with medicinal products that are substrates for OAT1/3 and BCRP transport may increase plasma concentrations of such medicinal products. Lumacaftor and ivacaftor are not inhibitors of OATP1B1, OATP1B3, and organic cation transporter (OCT) 1 and 2. Ivacaftor is not an inhibitor of OAT1 and OAT3.
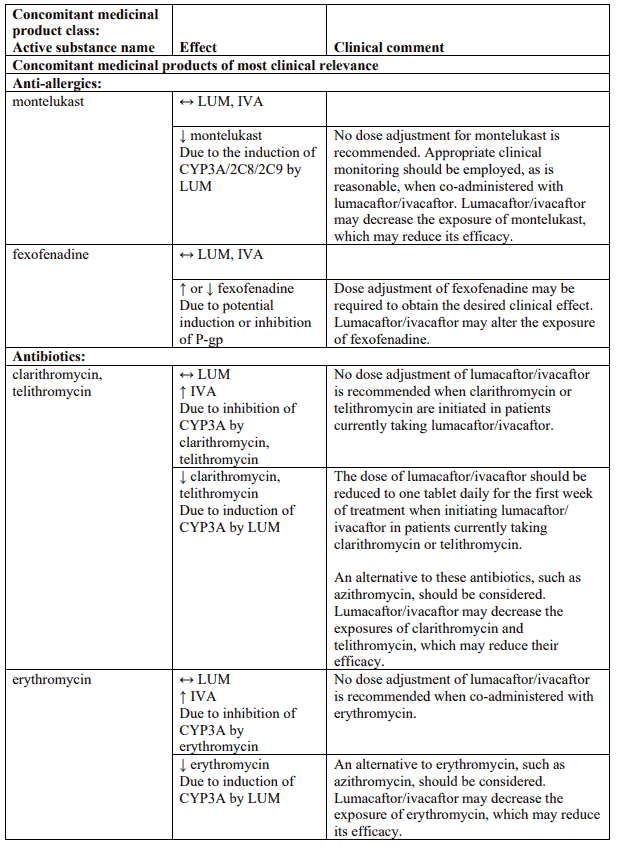
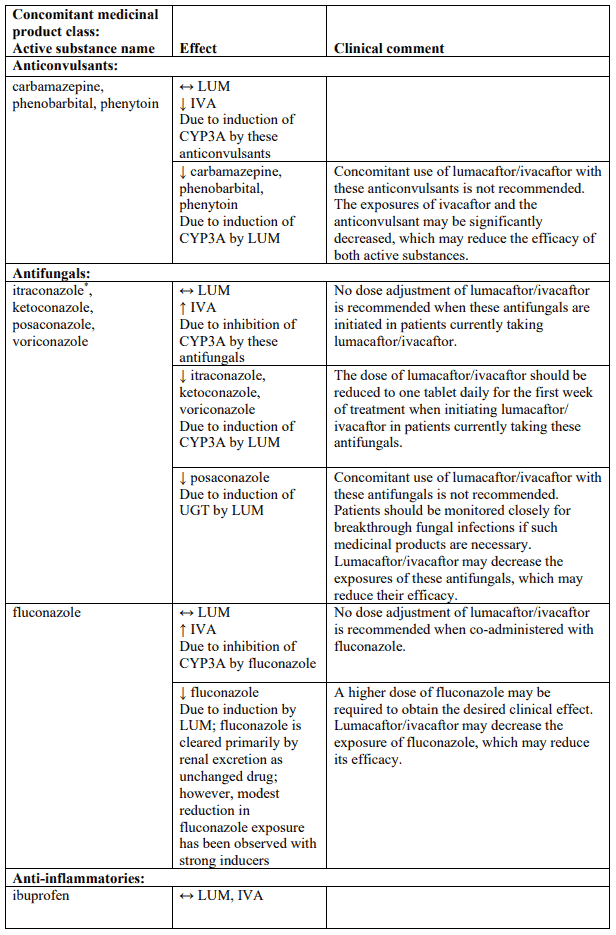
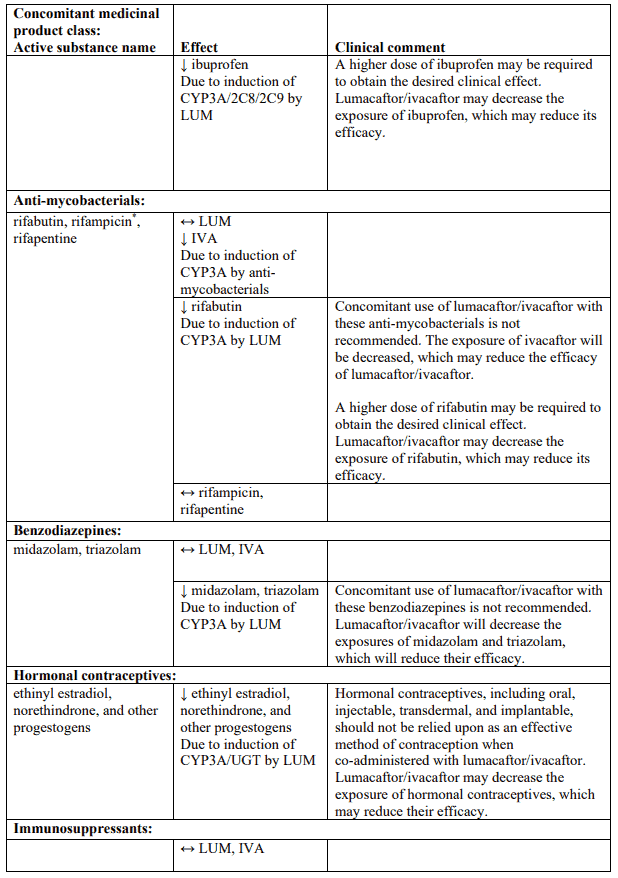
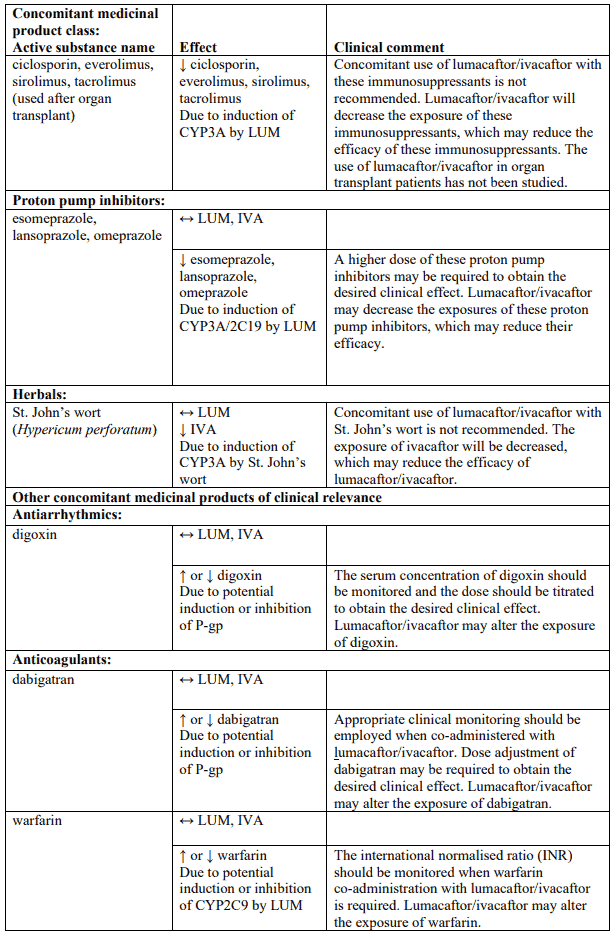
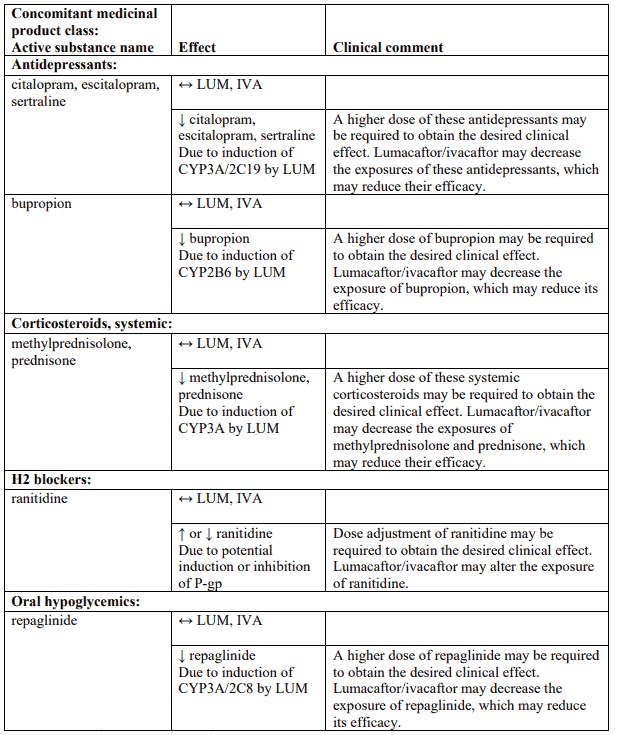
Table 4 provides the established or predicted effect of lumacaftor/ivacaftor on other medicinal products or the effect of other medicinal products on lumacaftor/ivacaftor. The information reported in Table 4 mostly derives from in vitro studies. The recommendations provided under “Clinical comment” in Table 4 are based on interaction studies, clinical relevance, or predicted interactions due to elimination pathways. Interactions that have the most clinical relevance are listed first.
Table 4. Established and other potentially significant interactions – dose recommendations for use of lumacaftor/ivacaftor with other medicinal products:
Note: ↑ = increase, ↓ = decrease, ↔ = no change; LUM = lumacaftor; IVA = ivacaftor. * Based on clinical interaction studies. All other interactions shown are predicted.
There have been reports of false positive urine screening tests for tetrahydrocannabinol (THC) in patients receiving Orkambi. An alternative confirmatory method should be considered to verify results.
Interaction studies have only been performed in adults
There are no or limited amount of data (less than 300 pregnancy outcomes) from the use of lumacaftor/ivacaftor in pregnant women. Animal studies with lumacaftor and ivacaftor do not indicate direct or indirect harmful effects with respect to developmental and reproductive toxicity, whereas effects were noted with ivacaftor only at maternally toxic doses (see section 5.3). As a precautionary measure, it is preferable to avoid the use of lumacaftor/ivacaftor during pregnancy unless the clinical condition of the mother requires treatment with lumacaftor/ivacaftor.
It is unknown whether lumacaftor and/or ivacaftor and metabolites are excreted in human milk. Available pharmacokinetic data in animals have shown excretion of both lumacaftor and ivacaftor into the milk of lactating female rats. As such, risks to the suckling child cannot be excluded. A decision must be made whether to discontinue breast-feeding or to discontinue/abstain from lumacaftor/ivacaftor therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
No human data on the effects of lumacaftor and/or ivacaftor on fertility are available. Lumacaftor had no effects on fertility and reproductive performance indices in male and female rats. Ivacaftor impaired fertility and reproductive performance indices in male and female rats (see section 5.3).
Ivacaftor, which is one of the active components of Orkambi, has a minor influence on the ability to drive and use machines. Ivacaftor may cause dizziness (see section 4.8).
Patients experiencing dizziness while taking Orkambi should be advised not to drive or use machines until symptoms abate.
The most common adverse reactions are dyspnoea (14.0%), diarrhoea (11.0%), and nausea (10.2%).
Serious adverse reactions included hepatobiliary events, e.g., transaminase elevations (0.5%), cholestatic hepatitis (0.3%) and hepatic encephalopathy (0.1%).
Adverse reactions identified from the 24-week, placebo-controlled, Phase 3 studies (trials 809-103 and 809-104) in patients aged 12 years and older and from a 24-week, placebo-controlled study in patients aged 6 to less than 12 years (trial 809-109), who are homozygous for the F508del mutation in the CFTR gene are presented in Table 5 and are listed by system organ class and frequency. Adverse reactions observed with ivacaftor alone are also provided in Table 5. Adverse reactions are ranked under the MedDRA frequency classification: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); and not known (frequency cannot be estimated using the available data).
Table 5. Adverse reactions in lumacaftor/ivacaftor-treated patients and in patients treated with ivacaftor alone:
| System organ class | Frequency | Adverse reactions |
|---|---|---|
| Infections and infestations | very common | Nasopharyngitis* |
| common | Upper respiratory tract infection, rhinitis | |
| Vascular disorders | uncommon | Hypertension |
| Nervous system disorders | very common | Headache, dizziness* |
| uncommon | Hepatic encephalopathy† | |
| Ear and labyrinth disorders | common | Ear pain*, ear discomfort*, tinnitus*, tympanic membrane hyperaemia*, vestibular disorder* |
| uncommon | Ear congestion* | |
| Respiratory, thoracic and mediastinal disorders | very common | Nasal congestion, dyspnoea, productive cough, sputum increased |
| common | Respiration abnormal, oropharyngeal pain, sinus congestion*, rhinorrhoea, pharyngeal erythema*, bronchospasm | |
| Gastrointestinal disorders | very common | Abdominal pain*, abdominal pain upper, diarrhoea, nausea |
| common | Flatulence, vomiting | |
| Hepatobiliary disorders | common | Transaminase elevations |
| uncommon | Cholestatic hepatitis‡ | |
| Skin and subcutaneous tissue disorders | common | Rash |
| Reproductive system and breast disorders | common | Menstruation irregular, dysmenorrhoea, metrorrhagia, breast mass* |
| uncommon | Menorrhagia, amenorrhoea, polymenorrhoea, breast inflammation*, gynaecomastia*, nipple disorder*, nipple pain*, oligomenorrhoea | |
| Investigations | very common | Bacteria in sputum* |
| common | Blood creatine phosphokinase increased | |
| uncommon | Blood pressure increased |
* Adverse reactions and frequencies observed in patients in clinical studies with ivacaftor monotherapy.
† 1 patient out of 738
‡ 2 patients out of 73
The safety data from 1,029 patients aged 12 years and older who were homozygous for the F508del mutation in the CFTR gene treated with lumacaftor/ivacaftor for up to an additional 96 weeks in the long-term safety and efficacy rollover study (trial 809-105) were similar to the 24-week, placebo-controlled studies (see section 5.1).
During trials 809-103 and 809-104, the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 x ULN was 0.8%, 2.0%, and 5.2%; and 0.5%, 1.9%, and 5.1% in lumacaftor/ivacaftorand placebo-treated patients, respectively. The incidence of transaminase-related adverse reactions was 5.1% and 4.6% in lumacaftor/ivacaftor-treated patients and those who received placebo, respectively. Seven patients who received lumacaftor/ivacaftor had liver-related serious adverse reactions with elevated transaminases, including 3 with concurrent elevation in total bilirubin. Following discontinuation of lumacaftor/ivacaftor, liver function tests returned to baseline or improved substantially in all patients (see section 4.4).
Among 7 patients with pre-existing cirrhosis and/or portal hypertension who received lumacaftor/ivacaftor in the placebo-controlled, Phase 3 studies, worsening liver function with increased ALT, AST, bilirubin, and hepatic encephalopathy was observed in one patient. The event occurred within 5 days of the start of dosing and resolved following discontinuation of lumacaftor/ivacaftor (see section 4.4).
Post–marketing cases of liver function decompensation including liver failure leading to death have been reported in CF patients with pre-existing cirrhosis with portal hypertension who were treated with lumacaftor/ivacaftor (see section 4.4).
During trials 809-103 and 809-104, the incidence of respiratory adverse reactions (e.g., chest discomfort, dyspnoea, bronchospasm, and respiration abnormal) was 26.3% in lumacaftor/ivacaftor-treated patients compared to 17.0% in patients who received placebo. The incidence of these adverse reactions was more common in patients with lower pre-treatment FEV1. Approximately three-quarters of the events began during the first week of treatment, and in most patients the events resolved without dosing interruption. The majority of adverse reactions were mild or moderate in severity, non-serious and did not result in treatment discontinuation (see section 4.4). During a 24-week, open-label, Phase 3b clinical study (trial 809-011 [Part B]) in 46 patients aged 12 years and older with advanced lung disease (ppFEV1 <40) [mean ppFEV1 29.1 at baseline (range: 18.3 to 42.0)], the incidence of respiratory adverse reactions was 65.2%. In the subgroup of 28 patients who were initiated at the full dose of lumacaftor/ivacaftor (2 tablets every 12 hours), the incidence was 71.4%, and in the 18 patients who were initiated at a reduced dose of lumacaftor/ivacaftor (1 tablet every 12 hours for up to 2 weeks, and subsequently increased to the full dose), the incidence was 55.6%. Of the patients who were initiated lumacaftor/ivacaftor at the full dose, one patient had a serious respiratory adverse reaction, three patients subsequently had their dose reduced, and three patients discontinued treatment. No serious respiratory adverse reactions, dose reductions or discontinuations were seen in patients who were initiated at the half dose (see section 4.4).
During trials 809-103 and 809-104, the incidence of combined menstrual abnormalities (amenorrhoea, dysmenorrhoea, menorrhagia, menstruation irregular, metrorrhagia, oligomenorrhoea, and polymenorrhoea) was 9.9% in lumacaftor/ivacaftor-treated female patients and 1.7% in placebo-treated females. These menstrual events occurred more frequently in the subset of female patients who were taking hormonal contraceptives (25.0%) versus patients who were not taking hormonal contraceptives (3.5%) (see section 4.5). Most of these reactions were mild or moderate in severity and non-serious. In lumacaftor/ivacaftor-treated patients, approximately two-thirds of these reactions resolved, and the median duration was 10 days.
During trials 809-103 and 809-104, adverse reactions related to increased blood pressure (e.g., hypertension, blood pressure increased) were reported in 0.9% (7/738) of patients treated with lumacaftor/ivacaftor and in no patients who received placebo.
In patients treated with lumacaftor/ivacaftor (mean baseline 114 mmHg systolic and 69 mmHg diastolic), the maximum increase from baseline in mean systolic and diastolic blood pressure was 3.1 mmHg and 1.8 mmHg, respectively. In patients who received placebo (mean baseline 114 mmHg systolic and 69 mmHg diastolic), the maximum increase from baseline in mean systolic and diastolic blood pressure was 0.9 mmHg and 0.9 mmHg, respectively.
The proportion of patients who experienced a systolic blood pressure value >140 mmHg or a diastolic blood pressure >90 mmHg on at least two occasions was 3.4% and 1.5% in patients treated with lumacaftor/ivacaftor, respectively, compared with 1.6% and 0.5% in patients who received placebo (see section 4.4).
The safety data of lumacaftor/ivacaftor were evaluated in 46 patients aged 1 to less than 2 years (trial 809-122), 60 patients aged 2 to 5 years (trial 809-115), 161 patients aged 6 to less than 12 years (trials 809-011 and 809-109) and in 194 patients aged 12 to 17 years with CF who are homozygous for the F508del mutation and who received lumacaftor/ivacaftor in clinical studies. Patients aged 12 to 17 years were included in trials 809-103 and 809-104.
The overall safety profile in these paediatric patients is generally consistent with that in adult patients.
Few selected adverse reactions are specifically reported in the paediatric population.
Long-term safety data from a 96-week rollover extension study (trial 809-116) in 57 patients aged 2 years and older who were homozygous for the F508del mutation in the CFTR gene were generally consistent with the 24-week parent study in patients aged 2 to 5 years (trial 809-115) and safety data in patients aged 6 to less than 12 years.
Long-term safety data from a 96-week rollover extension study in 239 patients aged 6 years and older who were homozygous for the F508del mutation in the CFTR gene (trial 809-110) were generally consistent with the 24-week parent studies in patients aged 6 to less than 12 years (trial 809-011 and trial 809-109).
During the 24-week, open-label Phase 3 clinical study in 58 patients aged 6 to less than 12 years (trial 809-011), the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 x ULN was 5.3%, 8.8%, and 19.3%. No patients had total bilirubin levels >2 x ULN.
Lumacaftor/ivacaftor dosing was maintained or successfully resumed after interruption in all patients with transaminase elevations, except 1 patient who discontinued treatment.
During the 24-week, placebo-controlled Phase 3 clinical study in 204 patients aged 6 to less than 12 years (trial 809-109), the incidence of maximum transaminase (ALT or AST) levels >8, >5, and >3 x ULN was 1.0%, 4.9%, and 12.6% in the lumacaftor/ivacaftor patients, and 2.0%, 3.0%, and 7.9% in the placebo-treated patients. No patients had total bilirubin levels >2 x ULN. Two patients in the lumacaftor/ivacaftor group and two patients in the placebo group discontinued treatment due to transaminase elevations.
During the 24-week, open-label Phase 3 clinical study (trial 809-011) in 58 patients aged 6 to less than 12 years (mean baseline ppFEV1 was 91.4), the incidence of respiratory adverse reactions was 6.9% (4/58).
During the 24-week, placebo-controlled Phase 3 clinical study (trial 809-109) in patients aged 6 to less than 12 years (mean baseline ppFEV1 was 89.8), the incidence of respiratory adverse reactions was 18.4% in lumacaftor/ivacaftor patients and 12.9% in placebo patients. A decline in ppFEV1 at initiation of therapy was observed during serial post dose spirometry assessments. The absolute change from pre-dose at 4 to6 hours post-dose was -7.7 on day 1 and -1.3 on day 15 in lumacaftor/ivacaftor patients. The post-dose decline was resolved by week 16.
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.