QTERN Film-coated tablet Ref.[7953] Active ingredients: Dapagliflozin Saxagliptin Saxagliptin and Dapagliflozin
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: AstraZeneca AB, SE-151 85, Södertälje, Sweden
Pharmacodynamic properties
Pharmacotherapeutic group: Drugs used in diabetes, combinations of oral blood glucose lowering drugs
ATC code: A10BD21
Mechanism of action
This medicinal product combines saxagliptin and dapagliflozin with complementary mechanisms of action to improve glycaemic control. Saxagliptin, through the selective inhibition of dipeptidyl peptidase-4 (DPP-4), enhances glucose-mediated insulin secretion (incretin effect). Dapagliflozin, a selective inhibitor of sodium-glucose co-transporter 2 (SGLT2), inhibits renal glucose reabsorption independently of insulin. Actions of both medicinal products are regulated by the plasma glucose level.
Saxagliptin is a highly potent (Ki: 1.3 nM), selective, reversible and competitive inhibitor of DPP-4, an enzyme responsible for the breakdown of incretin hormones. This results in a glucose-dependent increase in insulin secretion, thus reducing fasting and post-prandial blood glucose concentrations.
Dapagliflozin is a highly potent (Ki: 0.55 nM), selective and reversible inhibitor of sodium-glucose co-transporter 2 (SGLT2). Dapagliflozin blocks reabsorption of filtered glucose from the S1 segment of the renal tubule, effectively lowering blood glucose in a glucose dependent and insulin-independent manner. Dapagliflozin improves both fasting and post-prandial plasma glucose levels by reducing renal glucose reabsorption leading to urinary glucose excretion. The increased urinary glucose excretion with SGLT2 inhibition produces an osmotic diuresis, and can result in a reduction in systolic BP.
Pharmacodynamic effects
In patients with type 2 diabetes, administration of saxagliptin inhibited DPP-4 enzyme activity throughout a 24-hour period. The inhibition of plasma DPP-4 activity by saxagliptin for at least 24 hours after oral administration of saxagliptin is due to high potency, high affinity, and extended binding to the active site. After an oral glucose load, this produced in a 2- to 3-fold increase in circulating levels glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP), decreased glucagon concentrations, and increased beta-cell responsiveness, resulting in higher insulin and C-peptide concentrations. The rise in insulin from pancreatic beta-cells and the decrease in glucagon from pancreatic alpha-cells were associated with lower fasting glucose concentrations and reduced glucose excursion following an oral glucose load or a meal.
Dapagliflozin's glucuretic effect is observed after the first dose, is continuous over the 24-hour dosing interval, and is sustained for the duration of treatment. Increases in the amount of glucose excreted in the urine were observed in healthy subjects and in subjects with type 2 diabetes mellitus following the administration of dapagliflozin. Approximately 70 g of glucose was excreted in the urine per day (corresponding to 280 kcal/day) at a dapagliflozin dose of 10 mg/day in subjects with type 2 diabetes mellitus for 12 weeks. Evidence of sustained glucose excretion was seen in subjects with type 2 diabetes mellitus given dapagliflozin 10 mg/day for up to 2 years. Urinary uric acid excretion was also increased transiently (for 3-7 days) and accompanied by a sustained reduction in serum uric acid concentration. At 24 weeks, reductions in serum uric acid concentrations ranged from –48.3 to –18.3 micromoles/l (–0.87 to –0.33 mg/dl).
Clinical efficacy and safety
The safety and efficacy of the 5 mg saxagliptin/10 mg dapagliflozin fixed-dose combination was evaluated in three phase 3, randomised, double-blind, active/placebo-controlled clinical trials in 1169 adult subjects with type 2 diabetes mellitus. One trial with saxagliptin and dapagliflozin added concomitantly to metformin was conducted for 24 weeks. Two add-on therapy trials, which added either dapagliflozin to saxagliptin plus metformin or saxagliptin to dapagliflozin plus metformin, were also conducted for 24 weeks followed by a 28 week extension treatment period. The safety profile of the combined use of saxagliptin plus dapagliflozin in these trials for up to 52 weeks was comparable to the safety profiles for the mono-components.
Glycaemic control
Concomitant therapy with saxagliptin and dapagliflozin in patients inadequately controlled on metformin
A total of 534 adult patients with type 2 diabetes mellitus and inadequate glycaemic control on metformin alone (HbA1c ≥8% and ≤12%), participated in this 24-week randomised, double-blind, active comparator-controlled superiority trial to compare the combination of saxagliptin and dapagliflozin added concurrently to metformin, versus saxagliptin (DPP-4 inhibitor) or dapagliflozin (SGLT2 inhibitor) added to metformin. Patients were randomised to one of three double-blind treatment groups to receive saxagliptin 5 mg and dapagliflozin 10 mg added to metformin, saxagliptin 5 mg and placebo added to metformin, or dapagliflozin 10 mg and placebo added to metformin.
The saxagliptin and dapagliflozin group achieved significantly greater reductions in HbA1c versus either the saxagliptin group or dapagliflozin group at 24 weeks (see table 2).
Table 2. HbA1c at week 24 in active-controlled study comparing the combination of saxagliptin and dapagliflozin added concurrently to metformin with either saxagliptin or dapagliflozin added to metformin:
| Efficacy parameter | Saxagliptin 5 mg + dapagliflozin 10 mg + metformin N=1792 | Saxagliptin 5 mg + metformin N=1762 | Dapagliflozin 10 mg + metformin N=1792 |
|---|---|---|---|
| HbA1c (%) at week 241 | |||
| Baseline (mean) | 8.93 | 9.03 | 8.87 |
| Change from baseline (adjusted mean3) (95% Confidence interval [CI]) | −1.47 (−1.62, −1.31) | −0.88 (−1.03, −0.72) | −1.20 (−1.35, −1.04) |
| Difference from saxagliptin + metformin (adjusted mean3) (95% CI) | −0.594 (−0.81, −0.37) | - | - |
| Difference from dapagliflozin + metformin (adjusted mean3) (95% CI) | −0.275 (−0.48, −0.05) | - | - |
1 LRM = Longitudinal repeated measures (using values prior to rescue).
2 Randomised and treated patients with baseline and at least 1 post-baseline efficacy measurement.
3 Least squares mean adjusted for baseline value.
4 p-value <0.0001.
5 p-value=0.0166.
The majority of patients in this study had a baseline HbA1c of >8% (see table 3). The combination of saxagliptin and dapagliflozin added to metformin consistently demonstrated greater reductions in HbA1c irrespective of baseline HbA1c compared with saxagliptin or dapagliflozin alone added to metformin. In a separate pre-specified subgroup analysis, mean reductions from baseline in HbA1c were generally greater for patients with higher baseline HbA1c values.
Table 3. HbA1c subgroup analysis by baseline HbA1c at week 24 in randomised subjects:
| Treatments | Adjusted mean change from baseline by baseline HbA1c | ||
|---|---|---|---|
| <8.0% | ≥8% to <9.0% | ≥9.0% | |
| Saxagliptin + dapagliflozin + Metformin. Adjusted mean change from baseline (95% CI) | –0.80 (n=37) (–1.12, –0.47) | –1.17 (n=56) (–1.44, –0.90) | –2.03 (n=65) (–2.27, –1.80) |
| Saxagliptin + metformin Adjusted mean change from baseline (95% CI) | –0.69 (n=29) (–1.06, –0.33) | –0.51 (n=51) (–0.78, –0.25) | –1.32 (n=63) (–1.56, –1.09) |
| Dapagliflozin + metformin Adjusted mean change from baseline (95% CI) | –0.45 (n=37) (–0.77, –0.13) | –0.84 (n=52) (–1.11, –0.57) | –1.87 (n=62) (–2.11, –1.63) |
n = number of subjects with non-missing baseline and a Week 24 value.
Proportion of patients achieving HbA1c <7%
Forty-one point four percent (41.4%) (95% CI [34.5, 48.2]) of patients in the saxagliptin and dapagliflozin combination group achieved HbA1c levels of less than 7% compared to 18.3% (95% CI [13.0, 23.5]) patients in the saxagliptin group and 22.2% (95% CI [16.1, 28.3]) patients in the dapagliflozin group.
Add-on therapy with dapagliflozin in patients inadequately controlled on saxagliptin plus metformin
A 24-week randomised, double-blind, placebo-controlled study compared the sequential addition of 10 mg dapagliflozin to 5 mg saxagliptin and metformin to the addition of placebo to 5 mg saxagliptin (DPP-4 inhibitor) and metformin in patients with type 2 diabetes mellitus and inadequate glycaemic control (HbA1c ≥7% and ≤10.5%). Three hundred twenty (320) subjects were randomised equally into either the dapagliflozin added to saxagliptin plus metformin treatment group or placebo plus saxagliptin plus metformin treatment group. Patients who completed the initial 24-week study period were eligible to enter a controlled 28-week long-term study extension (52 weeks).
The group with dapagliflozin sequentially added to saxagliptin and metformin achieved statistically significant (p-value <0.0001) greater reductions in HbA1c versus the group with placebo sequentially added to saxagliptin plus metformin group at 24 weeks (see table 4). The effect in HbA1c observed at Week 24 was sustained at Week 52.
Add-on therapy with saxagliptin in patients inadequately controlled on dapagliflozin plus metformin
A 24-week randomised, double-blind, placebo-controlled study conducted on patients with type 2 diabetes mellitus and inadequate glycaemic control (HbA1c ≥7% and ≤10.5%) on metformin and dapagliflozin alone, compared the sequential addition of 5 mg saxagliptin to 10 mg dapagliflozin and metformin, to the addition of placebo to 10 mg dapagliflozin and metformin, 153 patients were randomised into the saxagliptin added to dapagliflozin plus metformin treatment group, and 162 patients were randomised into the placebo added to dapagliflozin plus metformin treatment group. Patients who completed the initial 24-week study period were eligible to enter a controlled 28 week long-term study extension (52 weeks). The safety profile of saxagliptin added to dapagliflozin plus metformin in the long-term treatment period was consistent with that previously observed in the clinical trial experience for the concomitant therapy study and that observed in the 24-week treatment period in this study.
The group with saxagliptin sequentially added to dapagliflozin and metformin achieved statistically significant (p-value <0.0001) greater reductions in HbA1c versus the group with placebo sequentially added to dapagliflozin plus metformin group at 24 weeks (see table 4). The effect in HbA1c observed at Week 24 was sustained at Week 52.
Table 4. HbA1c change from baseline at week 24 excluding data after rescue for randomised subjects – studies MB102129 and CV181168:
| Efficacy parameter | Sequential add-on clinical trials | |||
|---|---|---|---|---|
| Study MB102129 | Study CV181168 | |||
| Dapagliflozin 10 mg add to saxagliptin 5 mg + metformin (N=160)† | Placebo + saxagliptin 5 mg + metformin (N=160)† | Saxagliptin 5 mg added to dapagliflozin 10 mg + metformin (N=153)† | Placebo + dapagliflozin 10 mg + metformin (N=162)† | |
| HbA1c (%) at week 24* | ||||
| Baseline (mean) | 8.24 | 8.16 | 7.95 | 7.85 |
| Change from baseline (adjusted mean‡) (95% CI) | −0.82 (−0.96, 0.69) | −0.10 (−0.24, 0.04) | −0.51 (−0.63, −0.39) | −0.16 (−0.28, −0.04) |
| Difference in HbA1c effect Adjusted mean (95% CI) p-value | −0.72 (−0.91, −0.53) <0.0001 | −0.35 (−0.52, −0.18) <0.0001 | ||
* LRM = Longitudinal repeated measures (using values prior to rescue).
† N is the number of randomised and treated patients with baseline and at least 1 post-baseline efficacy measurement.
‡ Least squares mean adjusted for baseline value.
Proportion of patients achieving HbA1c <7%
The proportion of patients achieving HbA1c <7.0% at Week 24 in the add-on therapy with dapagliflozin to saxagliptin plus metformin trial was higher in the dapagliflozin plus saxagliptin plus metformin group 38.0% (95% CI [30.9, 45.1]) compared to the placebo plus saxagliptin plus metformin group 12.4% (95% CI [7.0, 17.9]). The effect in HbA1c observed at Week 24 was sustained at Week 52. The proportion of patients achieving HbA1c <7% at week 24 for add-on therapy with saxagliptin to dapagliflozin plus metformin trial was higher in the saxagliptin plus dapagliflozin plus metformin group 35.3% (95% CI [28.2, 42.2]) compared to the placebo plus dapagliflozin plus metformin group 23.1% (95% CI [16.9, 29.3]). The effect in HbA1c observed at Week 24 was sustained at Week 52.
Body weight
In the concomitant study, the adjusted mean change from baseline in body weight at week 24 (excluding data after rescue) was −2.05 kg (95% CI [−2.52, −1.58]) in the saxagliptin 5 mg plus dapagliflozin 10 mg plus metformin group and −2.39 kg (95% CI [−2.87, −1.91]) in the dapagliflozin 10 mg plus metformin group, while the saxagliptin 5 mg plus metformin group had no change (0.00 kg) (95% CI [−0.48, 0.49]).
Blood pressure
Treatment with the saxagliptin/dapagliflozin fixed dose combination resulted in change from baseline for systolic blood pressure ranging from –1.3 to –2.2 mmHg and for diastolic blood pressure ranging from –0.5 to –1.2 mmHg caused by its mild diuretic effect. The modest lowering effects on BP were consistent over time and a similar number of subjects had systolic BP <130 mmHg or diastolic BP < 80 mmHg at week 24 across the treatment groups.
Cardiovascular safety
In the pool of three studies, cardiovascular (CV) events that were adjudicated and confirmed as CV events were reported in a total of 1.0% of subjects in the saxagliptin plus dapagliflozin plus metformin group, 0.6% in the saxagliptin plus metformin group, and 0.9% in the dapagliflozin plus metformin group.
Cardiovascular outcomes studies in patients with type 2 diabetes mellitus
No cardiovascular outcomes studies have been conducted to evaluate the saxagliptin/dapagliflozin combination.
Saxagliptin assessment of vascular outcomes recorded in patients with diabetes mellitus - thrombolysis in myocardial infarction (SAVOR) study
SAVOR was a CV outcome trial in 16,492 patients with HbA1c ≥6.5% and <12% (12,959 with established CV disease; 3533 with multiple risk factors only) who were randomised to saxagliptin (n=8280) or placebo (n=8212) added to regional standards of care for HbA1c and CV risk factors. The study population included those ≥65 years (n=8561) and ≥75 years (n=2330), with normal or mild renal impairment (n=13,916) as well as moderate (n=2240) or severe (n=336) renal impairment.
The primary safety (non-inferiority) and efficacy (superiority) endpoint was a composite endpoint consisting of the time-to-first occurrence of any of the following major adverse CV events (MACE): CV death, nonfatal myocardial infarction, or nonfatal ischemic stroke.
After a mean follow up of 2 years, the trial met its primary safety endpoint demonstrating saxagliptin does not increase the cardiovascular risk in patients with type 2 diabetes compared to placebo when added to current background therapy.
No benefit was observed for MACE or all-cause mortality.
One component of the secondary composite endpoint, hospitalisation for heart failure, occurred at a greater rate in the saxagliptin group (3.5%) compared with the placebo group (2.8%), with nominal statistical significance favouring placebo [HR=1.27; (95% CI 1.07, 1.51); P=0.007]. Clinically relevant factors predictive of increased relative risk with saxagliptin treatment could not be definitively identified. Subjects at higher risk for hospitalisation for heart failure, irrespective of treatment assignment, could be identified by known risk factors for heart failure such as baseline history of heart failure or impaired renal function. However, subjects on saxagliptin with a history of heart failure or impaired renal function at baseline were not at an increased risk relative to placebo for the primary or secondary composite endpoints or all-cause mortality.
Another secondary endpoint, all-cause mortality, occurred at a rate of 5.1% in the saxagliptin group and 4.6% in the placebo group. CV deaths were balanced across the treatment groups. There was a numerical imbalance in non-CV death, with more events on saxagliptin (1.8%) than placebo (1.4%) [HR=1.27; (95% CI 1.00, 1.62); P=0.051].
Dapagliflozin Effect on Cardiovascular Events (DECLARE)
Dapagliflozin Effect on Cardiovascular Events (DECLARE) was an international, multicentre, randomised, double-blind, placebo-controlled clinical study conducted to determine the effect of dapagliflozin compared with placebo on cardiovascular outcomes when added to current background therapy. All patients had type 2 diabetes mellitus and either at least two additional cardiovascular risk factors (age ≥55 years in men or ≥60 years in women and one or more of dyslipidaemia, hypertensionor current tobacco use) or established cardiovascular disease.
Of 17 160 randomised patients, 6 974 (40.6%) had established cardiovascular disease and 10 186 (59.4%) did not have established cardiovascular disease. 8 582 patients were randomised to dapagliflozin 10 mg and 8 578 to placebo, and were followed for a median of 4.2 years.
The mean age of the study population was 63.9 years, 37.4% were female. In total, 22.4% had had diabetes for ≤5 years, mean duration of diabetes was 11.9 years. Mean HbA1c was 8.3% and mean BMI was 32.1 kg/m².
At baseline, 10.0% of patients had a history of heart failure. Mean eGFR was 85.2 mL/min/1.73 m², 7.4% of patients had eGFR <60 mL/min/1.73 m², and 30.3% of patients had micro- or macroalbuminuria (urine albumin to creatinine ratio [UACR] ≥30 to ≤300 mg/g or >300 mg/g, respectively).
Most patients (98%) used one or more diabetic medications at baseline, including metformin (82%), insulin (41%) and sulfonylurea (43%).
The primary endpoints were time to first event of the composite of cardiovascular death, myocardial infarction or ischaemic stroke (MACE) and time to first event of the composite of hospitalisation for heart failure or cardiovascular death. The secondary endpoints were a renal composite endpoint and all-cause mortality.
Major adverse cardiovascular events:
Dapagliflozin 10 mg demonstrated non-inferiority versus placebo for the composite of cardiovascular death, myocardial infarction or ischaemic stroke (one-sided p<0.001).
Heart failure or cardiovascular death:
Dapagliflozin 10 mg demonstrated superiority versus placebo in preventing the composite of hospitalisation for heart failure or cardiovascular death (Figure 1). The difference in treatment effect was driven by hospitalisation for heart failure, with no difference in cardiovascular death (Figure 2).
The treatment benefit of dapagliflozin over placebo was observed both in patients with and without established cardiovascular disease, with and without heart failure at baseline, and was consistent across key subgroups, including age, gender, renal function (eGFR) and region.
Figure 1. Time to first occurrence of hospitalisation for heart failure or cardiovascular death:
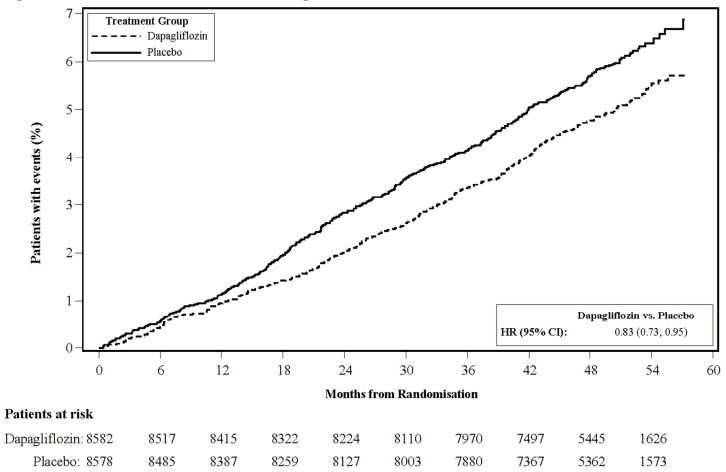
Patients at risk is the number of patients at risk at the beginning of the period.
HR = Hazard ratio CI=Confidence interval.
Results on primary and secondary endpoints are displayed in Figure 2. Superiority of dapagliflozin over placebo was not demonstrated for MACE (p=0.172). The renal composite endpoint and all-cause mortality were therefore not tested as part of the confirmatory testing procedure.
Figure 2. Treatment effects for the primary composite endpoints and their components, and the secondary endpoints and components:
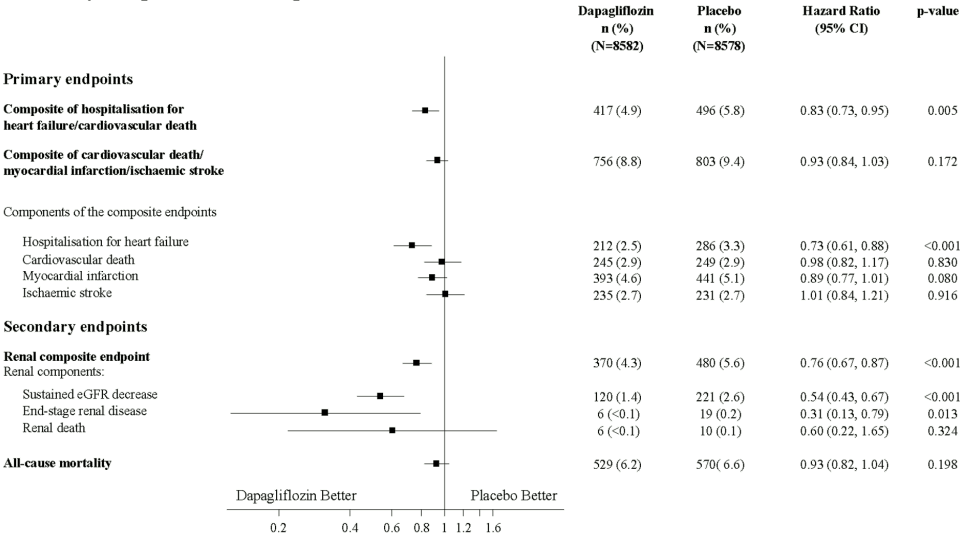
Renal composite endpoint defined as: sustained confirmed ≥40% decrease in eGFR to eGFR <60 mL/min/1.73 m² and/or end-stage renal disease (dialysis ≥90 days or kidney transplantation, sustained confirmed eGFR <15 mL/min/1.73 m²) and/or renal or cardiovascular death.
p-values are two-sided. p-values for the secondary endpoints and for single components are nominal. Time to first event was analysed in a Cox proportional hazards model. The number of first events for the single components are the actual number of first events for each component and does not add up to the number of events in the composite endpoint.
CI = confidence interval.
Nephropathy:
Dapagliflozin reduced the incidence of events of the composite of confirmed sustained eGFR decrease, end-stage renal disease, renal or cardiovascular death. The difference between groups was driven by reductions in events of the renal components; sustained eGFR decrease, end-stage renal disease and renal death (Figure 2).
The hazard ratio for time to nephropathy (sustained eGFR decrease, end-stage renal disease and renal death) was 0.53 (95% CI 0.43, 0.66) for dapagliflozin versus placebo.
In addition, dapagliflozin reduced the new onset of sustained albuminuria (hazard ratio 0.79 [95% CI 0.72, 0.87]) and led to greater regression of macroalbuminuria (hazard ratio 1.82 [95% CI 1.51, 2.20]) compared with placebo.
Renal impairment
Moderate renal impairment CKD 3A (eGFR ≥45 to <60 mL/min/1.73 m²)
Dapagliflozin:
The efficacy of dapagliflozin was assessed in a dedicated study in diabetic patients with an eGFR ≥45 to <60 mL/min/1.73 m² who had inadequate glycaemic control on usual care. Treatment with dapagliflozin resulted in reductions in HbA1c and body weight compared with placebo (Table 5).
Table 5. Results at week 24 of a placebo-controlled study of dapagliflozin in diabetic patients with an eGFR ≥45 to <60 mL/min/1.73 m²:
| Dapagliflozina 10 mg | Placeboa | |
|---|---|---|
| Nb | 159 | 161 |
| HbA1c (%) | ||
| Baseline (mean) | 8.35 | 8.03 |
| Change from baselineb | −0.37 | −0.03 |
| Difference from placebob (95% CI) | −0.34* (−0.53, −0.15) | |
| Body weight (kg) | ||
| Baseline (mean) | 92.51 | 88.30 |
| Percent change from baselinec | −3.42 | −2.02 |
| Difference in percent change from placeboc (95% CI) | −1.43* (−2.15, −0.69) | |
a Metformin or metformin hydrochloride were part of the usual care in 69.4% and 64.0% of the patients for the dapagliflozin and placebo groups, respectively.
b Least squares mean adjusted for baseline value
c Derived from least squares mean adjusted for baseline value
* p≤0.001
At week 24, treatment with dapagliflozin demonstrated reductions in fasting plasma glucose (FPG) −1.19 mmol/L (−21.46 mg/dL) compared to −0.27 mmol/L (−4.87 mg/dL) for placebo (p≤0.001), and reductions in seated systolic blood pressure (SBP) −4.8 mmHg compared to −1.7 mmHg for placebo (p<0.05).
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Qtern in all subsets of the paediatric population in the treatment of type 2 diabetes (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Saxagliptin/dapagliflozin combination
Overall, the pharmacokinetics of saxagliptin and dapagliflozin were not affected in clinically relevant manner when administered as a fixed dose combination compared with independent doses of saxagliptin and dapagliflozin.
The following reflects the pharmacokinetic properties of the saxagliptin/dapagliflozin fixed dose combination unless stated that the presented data are from administration of saxagliptin or dapagliflozin.
Bioequivalence has been confirmed between the Qtern 5 mg/10 mg tablet and the individual saxagliptin 5 mg and dapagliflozin 10 mg tablets after single dose administration in the fasted state in healthy subjects. The pharmacokinetics of dapagliflozin, and saxagliptin and its major metabolite were similar in healthy subjects and in patients with type 2 diabetes.
Administration of the saxagliptin/dapagliflozin fixed dose combination with a high-fat meal decreases dapagliflozin Cmax by up to 35% and prolongs Tmax by approximately 1.5 hours, but does not alter AUC as compared with the fasted state. These changes are not considered to be clinically meaningful. There was no food effect observed for saxagliptin. This medicinal product can be administered with or without food.
Drug interactions
Saxagliptin/dapagliflozin combination
No interaction studies have been performed with the saxagliptin/dapagliflozin fixed dose combination and other medicinal products. Such studies have been conducted with the individual active substances.
Saxagliptin
In in vitro studies, saxagliptin and its major metabolite neither inhibited CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, or 3A4, nor induced CYP1A2, 2B6, 2C9, or 3A4.
Dapagliflozin
In in vitro studies, dapagliflozin neither inhibited cytochrome P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, nor induced CYP1A2, CYP2B6 or CYP3A4. Therefore, dapagliflozin is not expected to alter the metabolic clearance of coadministered medicinal products that are metabolised by these enzymes.
Absorption
Saxagliptin
Saxagliptin was rapidly absorbed after oral administration in the fasted state, with maximum plasma concentrations (Cmax) of saxagliptin and its major metabolite attained within 2 and 4 hours (Tmax), respectively. The Cmax and AUC values of saxagliptin and its major metabolite increased proportionally with the increment in the saxagliptin dose, and this dose-proportionality was observed in doses up to 400 mg. Following a 5 mg single oral dose of saxagliptin to healthy subjects, the mean plasma AUC values for saxagliptin and its major metabolite were 78 ng h/mL and 214 ng h/mL, respectively. The corresponding plasma Cmax values were 24 ng/mL and 47 ng/mL, respectively. The intra-subject coefficients of variation for saxagliptin Cmax and AUC were less than 12%.
Dapagliflozin
Dapagliflozin was rapidly and well absorbed after oral administration. Maximum dapagliflozin plasma concentrations (Cmax) were usually attained within 2 hours after administration in the fasted state. Geometric mean steady-state dapagliflozin Cmax and AUCτ values following once daily 10 mg doses of dapagliflozin were 158 ng/mL and 628 ng h/mL, respectively. The absolute oral bioavailability of dapagliflozin following the administration of a 10 mg dose is 78%.
Distribution
Saxagliptin
The in vitro protein binding of saxagliptin and its major metabolite in human serum is negligible. Thus, changes in blood protein levels in various disease states (e.g. renal or hepatic impairment) are not expected to alter the disposition of saxagliptin. The volume of distribution of saxagliptin was 205 L.
Dapagliflozin
Dapagliflozin is approximately 91% protein bound. Protein binding was not altered in various disease states (e.g. renal or hepatic impairment). The mean steady-state volume of distribution of dapagliflozin was 118 L.
Biotransformation
Saxagliptin
The biotransformation of saxagliptin is primarily mediated by cytochrome P450 3A4/5 (CYP3A4/5). The major active metabolite of saxagliptin, 5-OH-saxagliptin, is also a selective, reversible, competitive DPP-4 inhibitor, half as potent as saxagliptin.
Dapagliflozin
Dapagliflozin is extensively metabolised, primarily to yield dapagliflozin 3-O-glucuronide, which is an inactive metabolite. Dapagliflozin 3-O-glucuronide or other metabolites do not contribute to the glucose-lowering effects. The formation of dapagliflozin 3-O-glucuronide is mediated by UGT1A9, an enzyme present in the liver and kidney, and CYP-mediated metabolism was a minor clearance pathway in humans.
Elimination
Saxagliptin
The mean plasma terminal half-life (t1/2) values for saxagliptin and its major metabolite are 2.5 hours and 3.1 hours respectively, and the mean t1/2 value for plasma DPP-4 inhibition was 26.9 hours. Saxagliptin is eliminated by both renal and hepatic pathways. Following a single 50 mg dose of 14C-saxagliptin, 24%, 36%, and 75% of the dose was excreted in the urine as saxagliptin, its active metabolite, and total radioactivity, respectively. The average renal clearance of saxagliptin (~230 mL/min) was greater than the average estimated glomerular filtration rate (~120 mL/min), suggesting some active renal excretion.
Dapagliflozin
The mean plasma terminal half-life (t1/2) for dapagliflozin was 12.9 hours following a single oral dose of dapagliflozin 10 mg to healthy subjects. The mean total systemic clearance of dapagliflozin administered intravenously was 207 mL/min. Dapagliflozin and related metabolites are primarily eliminated via urinary excretion with less than 2% as unchanged dapagliflozin.
Linearity
Saxagliptin
The Cmax and AUC of saxagliptin and its major metabolite increased proportionally to the saxagliptin dose. No appreciable accumulation of either saxagliptin or its major metabolite was observed with repeated once-daily dosing at any dose level. No dose- and time-dependence was observed in the clearance of saxagliptin and its major metabolite over 14 days of once-daily dosing with saxagliptin at doses ranging from 2.5 mg to 400 mg.
Dapagliflozin
Dapagliflozin exposure increased proportional to the increment in dapagliflozin dose over the range of 0.1 to 500 mg and its pharmacokinetics did not change with time upon repeated daily dosing for up to 24 weeks.
Special populations
Renal impairment
Saxagliptin: After a single dose of saxagliptin in subjects with mild, moderate or severe renal impairment (or ESRD) classified on the basis of creatinine clearance the mean AUC values of saxagliptin were 1.2-, and up to 2.1- and 4.5- fold higher, respectively, than AUC values in subjects with normal renal function. The AUC values of 5-OH-saxagliptin were also increased. The degree of renal impairment did not affect the Cmax of saxagliptin or its major metabolite. D Dapagliflozin: At steady-state (20 mg once-daily dapagliflozin for 7 days), subjects with type 2 diabetes mellitus and mild, moderate or severe renal impairment (as determined by iohexol plasma clearance) had mean systemic exposures of dapagliflozin of 32%, 60% and 87% higher, respectively, than those of subjects with type 2 diabetes mellitus and normal renal function. The steady-state 24-hour urinary glucose excretion was highly dependent on renal function and 85, 52, 18 and 11 g of glucose/day was excreted by subjects with type 2 diabetes mellitus and normal renal function or mild, moderate or severe renal impairment, respectively. The impact of haemodialysis on dapagliflozin exposure is not known.
Hepatic impairment
Saxagliptin: In subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B), or severe (Child-Pugh class C) hepatic impairment the exposures to saxagliptin were 1.1-, 1.4- and 1.8-fold higher, respectively, and the exposures to BMS-510849 (saxagliptin metabolite) were 22%, 7% and 33% lower, respectively, than those observed in healthy subjects.
Dapagliflozin: In subjects with mild or moderate hepatic impairment (Child-Pugh classes A and B), mean Cmax and AUC of dapagliflozin were up to 12% and 36% higher, respectively, compared to healthy matched control subjects. These differences were not considered to be clinically meaningful. In subjects with severe hepatic impairment (Child-Pugh class C) mean Cmax and AUC of dapagliflozin were 40% and 67% higher than matched healthy controls, respectively.
Elderly
Saxagliptin: Elderly patients (65–80 years) had about 60% higher saxagliptin AUC than young patients (18–40 years). This is not considered clinically meaningful, therefore, no dose adjustment for saxagliptin is recommended on the basis of age alone.
Dapagliflozin: There is no clinically meaningful increase in exposure based on age alone in subjects up to 70 years old. However, an increased exposure due to age-related decrease in renal function can be expected. There are insufficient data to draw conclusions regarding exposure in patients >70 years old.
Gender
Saxagliptin: Females had approximately 25% higher systemic exposure values for saxagliptin. There were no clinically relevant differences observed in saxagliptin pharmacokinetics between males and females.
Dapagliflozin: The mean dapagliflozin AUCss in females was estimated to be about 22% higher than in males.
Race
Saxagliptin: Race was not identified as a statistically significant covariate on the apparent clearance of saxagliptin and its metabolite.
Dapagliflozin: There were no clinically relevant differences in systemic exposures between White, Black or Asian races.
Body weight
Saxagliptin: Body weight had a small and non-clinically meaningful impact on saxagliptin exposure. Females had approximately 25% higher systemic-exposure values for saxagliptin, this difference is considered not clinically relevant.
Dapagliflozin: Dapagliflozin exposure was found to decrease with increased weight. Consequently, low-weight patients may have somewhat increased exposure and patients with high-weight somewhat decreased exposure. However, the differences in exposure were not considered clinically meaningful.
Preclinical safety data
Non-clinical studies of either saxagliptin or dapagliflozin revealed no special hazard for humans based on conventional studies of safety pharmacology, genotoxicity or carcinogenicity.
Saxagliptin produced reversible skin lesions (scabs, ulcerations and necrosis) in extremities (tail, digits, scrotum and/or nose) in cynomolgus monkeys. The no effect level (NOEL) for the lesions is 1 and 2 times the human exposure of saxagliptin and the major metabolite respectively, at the recommended human dose (RHD) of 5 mg/day. The clinical relevance of the skin lesions is not known and skin lesions have not been observed in humans.
Immune related findings of minimal, nonprogressive, lymphoid hyperplasia in spleen, lymph nodes and bone marrow with no adverse sequelae have been reported in all species tested at exposures starting from 7 times the RHD.
Saxagliptin produced gastrointestinal toxicity in dogs, including bloody/mucoid faeces and enteropathy at higher doses with a NOEL 4 and 2 times the human exposure for saxagliptin and the major metabolite, respectively at RHD. The effect on offspring body weights were noted until postnatal day 92 and 120 in females and males, respectively.
Reproductive and developmental toxicity
Saxagliptin has effects on fertility in male and female rats at high doses producing overt signs of toxicity. Saxagliptin was not teratogenic at any doses evaluated in rats or rabbits. At high doses in rats, saxagliptin caused reduced ossification (a developmental delay) of the foetal pelvis and decreased foetal body weight (in the presence of maternal toxicity), with a NOEL 303 and 30 times the human exposure for saxagliptin and the major metabolite, respectively, at RHD. In rabbits, the effects of saxagliptin were limited to minor skeletal variations observed only at maternally toxic doses (NOEL 158 and 224 times the human exposure for saxagliptin and the major metabolite, respectively at RHD). In a pre- and postnatal developmental study in rats, saxagliptin caused decreased pup weight at maternally toxic doses, with NOEL 488 and 45 times the human exposure for saxagliptin and the major metabolite, respectively at RHD. The effect on offspring body weights were noted until postnatal day 92 and 120 in females and males, respectively.
Direct administration of dapagliflozin to weanling juvenile rats and indirect exposure during late pregnancy (corresponding to the second and third trimesters of pregnancy with respect to human renal maturation) and lactation are each associated with increased incidence and/or severity of renal pelvic and tubular dilatations in progeny.
In a juvenile study, when dapagliflozin was dosed directly to young rats from postnatal day 21 until postnatal day 90, renal pelvic and tubular dilatations (with dose-related increases in kidney weight and macroscopic kidney enlargement) were reported at all dose levels; pup exposures at the lowest dose tested were ≥15 times the maximum recommended human dose. The renal pelvic and tubular dilatations observed in juvenile animals did not fully reverse within the approximate 1-month recovery period.
Dapagliflozin dosed to maternal rats from gestation day 6 through postnatal day 21, and pups were indirectly exposed in utero and throughout lactation. Increased incidence or severity of renal pelvic dilatation was observed in adult offspring of treated dams, although only at the highest dose tested (at maternal and pup dapagliflozin exposures of 1 415 times and 137 times, respectively, the human values at the maximum recommended human dose [MRHD]). Additional developmental toxicity was limited to dose-related reductions in pup body weights, and observed only at doses ≥15 mg/kg/day (pup exposures ≥29 times the human values at the MRHD). Maternal toxicity was evident only at the highest dose tested, and limited to transient reductions in body weight and food consumption at dose. The NOAEL for developmental toxicity is associated with a maternal systemic exposure 19 times the human values at the MRHD.
In studies of embryo-foetal development in rabbits, dapagliflozin caused neither maternal nor developmental toxicities at any dose tested; the highest dose tested corresponded to a systemic exposure 1191 times the MRHD. In rats, dapagliflozin was neither embryolethal nor teratogenic at exposures up to 1441 times the human values at the MRHD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.