RHAPSIDO Film-coated tablet Ref.[116560] Active ingredients: Remibrutinib
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, selective immunosuppressants
ATC code: L04AA60
Mechanism of action
Remibrutinib is a selective Bruton's tyrosine kinase (BTK) inhibitor that forms a covalent bond with a cysteine residue in the BTK active site, leading to durable inactivation of BTK. The therapeutic effect of remibrutinib in CSU is achieved through inhibition of mast cell and basophil degranulation, including release of histamine and other proinflammatory mediators, mediated by pathogenic IgE or IgG directed against the FcεR1 or IgE.
Pharmacodynamic effects
Cardiac electrophysiology
The effects of remibrutinib on QTc interval prolongation were predicted using concentration-QTc analysis. The upper bound of the 90% confidence interval for the predicted mean change in QTcF was below 10 msec at the expected Cmax at supratherapeutic exposures. Therefore, no clinically significant prolongation of QTcF interval is expected with therapeutic dosing of remibrutinib.
Clinical efficacy and safety
The efficacy and safety of remibrutinib were evaluated in two identical, multicentre, randomised, double-blind, placebo-controlled phase III studies (REMIX-1 and REMIX-2) in adult patients with inadequately controlled CSU despite treatment with second-generation H1 antihistamines.
In REMIX-1 and REMIX-2, patients were randomised in a 2:1 ratio to receive either remibrutinib 25 mg or placebo, respectively, twice daily via the oral route for 24 weeks during the double-blind treatment period and continued in a 28-week open-label treatment period during which all patients received remibrutinib 25 mg twice daily.
REMIX-1 and REMIX-2 enrolled a total of 925 adult patients diagnosed with CSU that was inadequately controlled despite treatment with a standard dose of a second-generation H1 antihistamine as defined by the presence of itch and hives for ≥6 consecutive weeks. All patients were required to have a weekly urticaria activity score (UAS7) ≥16 (range 0 to 42), a weekly itch severity score (ISS7) ≥6 (range 0 to 21) and a weekly hives severity score (HSS7) ≥6 (range 0 to 21) for 7 days prior to randomisation. In addition to all patients receiving a stable dose of a second-generation H1 antihistamine (background therapy), patients were allowed to use another second-generation H1 antihistamine on an "as-needed" basis (rescue therapy) in doses up to 4-fold the standard dose. Patients were excluded from these studies if they had evidence of clinically significant cardiovascular disease, a significant bleeding risk, coagulation disorders, ongoing, chronic or recurrent infection, chronic or acute hepatic disease with evidence of ongoing hepatitis C or B, history of renal disease, history of gastrointestinal bleeding or history of malignancy in the last 5 years.
Demographics and baseline characteristics were generally well balanced across all groups. In REMIX-1 and REMIX-2, the median age was 45 years (range: 18-79 years) and 41 years (range: 18-81 years), with 9.6% and 7.7% ≥65 years of age and 68.3% and 65.3% female patients, respectively. Patients had a mean UAS7 of 30.28 and 29.99, a mean ISS7 of 14.59 and 14.15, and a mean HSS7 of 15.69 and 15.84, respectively. At baseline, 63.4% and 59.1% of the patients had severe disease (UAS7 ≥28) and 35.1% and 38.7% had moderate disease (UAS7 >16 and <28), respectively. 51.7% and 46.6% of the patients had previous experience of angioedema in REMIX-1 and REMIX-2, respectively. 68.1% and 69.2% of patients were anti-IgE biologic naive in REMIX-1 and REMIX-2, respectively. The most common prior anti-IgE biologic used was omalizumab (19.5% and 19.0% in REMIX-1 and REMIX-2, respectively).
The reported mean duration of CSU at enrollment across treatment groups was 6.6 and 5.2 years in REMIX-1 and REMIX-2, respectively, with 39.4% and 29.5% of patients having had a duration of CSU >5 years.
The primary endpoint for the pivotal studies was:
- absolute change from baseline in UAS7 at week 12.
The secondary endpoints for the pivotal studies were:
- absolute change from baseline in ISS7 and HSS7 at week 12
- proportion of patients who achieved well-controlled disease (UAS7 ≤6) at weeks 2 and 12
- proportion of patients who achieved complete absence of itch and hives (UAS7 = 0) at week 12
- proportion of patients who achieved Dermatology Life Quality Index (DLQI) score = 0-1 (yes/no) at week 12
- number of weeks with sustained disease activity control (UAS7 ≤6) up to week 12
- number of angioedema-free weeks (weekly angioedema activity score [AAS7] = 0) up to week 12.
Clinical response
In both REMIX-1 and REMIX-2, the primary and all secondary endpoints were met and showed statistically significant and clinically meaningful improvements in itch and hives symptoms in patients treated with remibrutinib compared to patients given placebo. Results are presented in Table 2 and Figure 1.
Table 2. Efficacy results in REMIX-1 and REMIX-2 at week 12a,b:
| REMIX-1 | REMIX-2 | |||
| Remibrutinib (N=309) | Placebo (N=153) | Remibrutinib (N=297) | Placebo (N=153) | |
| Change from baseline in UAS7 at week 12 | ||||
| LS mean (SE) CFB | -20.02 (0.716) | -13.79 (0.980) | -19.41 (0.702) | -11.73 (0.948) |
| LS mean (SE) CFB difference vs placebo | -6.22 (1.136) | -7.68 (1.136) | ||
| 95% CI for difference | -8.45, -4.00 | -9.91, -5.46 | ||
| p-value | <0.001 | <0.001 | ||
| Change from baseline in ISS7 at week 12 | ||||
| LS mean (SE) CFB | -9.52 (0.343) | -6.89 (0.470) | -8.95 (0.335) | -5.72 (0.454) |
| LS mean (SE) CFB difference vs placebo | -2.63 (0.544) | -3.23 (0.545) | ||
| 95% CI for difference | -3.70, -1.56 | -4.29, -2.16 | ||
| p-value | <0.001 | <0.001 | ||
| Change from baseline in HSS7 at week 12 | ||||
| LS mean (SE) CFB | -10.47 (0.401) | -6.86 (0.548) | -10.47 (0.394) | -6.00 (0.531) |
| LS mean (SE) CFB difference vs placebo | -3.61 (0.635) | -4.47 (0.634) | ||
| 95% CI for difference | -4.85, -2.36 | -5.71, -3.23 | ||
| p-value | <0.001 | <0.001 | ||
| Proportion of patients with UAS7 ≤6 at week 2 | ||||
| n (%) | 104 (33.7) | 5 (3.3) | 89 (30.0) | 9 (5.9) |
| Treatment difference vs placebo | 30.20 | 24.55 | ||
| (95% CI) | 24.30, 36.10 | 18.31, 30.80 | ||
| p-value | <0.001 | <0.001 | ||
| Proportion of patients with UAS7 ≤6 at week 12 | ||||
| n (%) | 154 (49.8) | 38 (24.8) | 139 (46.8) | 30 (19.6) |
| Treatment difference vs placebo | 25.44 | 27.61 | ||
| (95% CI) | 16.48, 34.39 | 19.14, 36.08 | ||
| p-value | <0.001 | <0.001 | ||
| Proportion of patients with UAS7 = 0 at week 12 | ||||
| n (%) | 96 (31.1) | 16 (10.5) | 83 (27.9) | 10 (6.5) |
| Treatment difference vs placebo | 20.55 | 21.60 | ||
| (95% CI) | 13.35, 27.75 | 15.10, 28.10 | ||
| p-value | <0.001 | <0.001 | ||
| Proportion of patients with DLQI = 0-1 response at week 12 | ||||
| n (%) | 120 (39.0) | 34 (22.2) | 106 (35.7) | 28 (18.3) |
| Treatment difference vs placebo | 17.65 | 18.21 | ||
| (95% CI) | 9.14, 26.16 | 9.96, 26.45 | ||
| p-value | <0.001 | <0.001 | ||
| Cumulative number of weeks with UAS7 ≤6 between baseline and week 12 | ||||
| LS mean (SE) | 5.17 (0.414) | 1.92 (0.241) | 4.50 (0.464) | 1.38 (0.216) |
| Rate ratio | 2.69 | 3.26 | ||
| (95% CI) | (2.01, 3.61) | (2.26, 4.71) | ||
| p-value | <0.001 | <0.001 | ||
| Cumulative number of weeks with AAS7 = 0 between baseline and week 12 | ||||
| LS mean (SE) | 8.43 (0.274) | 6.72 (0.330) | 8.81 (0.308) | 6.68 (0.343) |
| Rate ratio | 1.25 | 1.32 | ||
| (95% CI) | (1.12, 1.41) | (1.17, 1.49) | ||
| p-value | <0.001 | <0.001 | ||
LS mean: Least squares mean, SE: standard error, CFB: change from baseline, CI: confidence interval, p-value: one-sided p-value, UAS7: weekly urticaria activity score, ISS7 score: weekly itch severity score, HSS7: weekly hive severity score, DLQI: dermatology life quality index, AAS7: weekly angioedema activity score.
a All endpoints with nominal one-sided p<0.001
b One endpoint from week 2 (all other endpoints are from week 12)
Figure 1. Mean change from baseline in UAS7 up to week 12 in REMIX-1 and REMIX-2 (observed data):
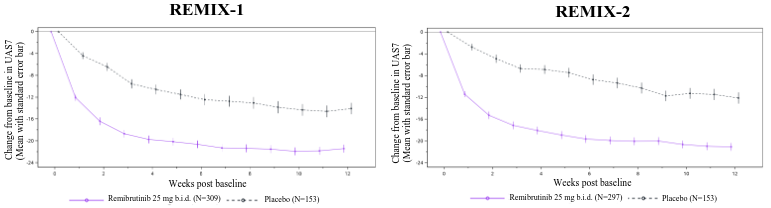
b.i.d. = twice daily
Subgroup analyses demonstrated a consistent treatment benefit with remibrutinib over placebo across subgroups including prior exposure to anti-IgE biologics and total IgE level.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Rhapsido in one or more subsets of the paediatric population in CSU (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Remibrutinib is rapidly absorbed and reaches Cmax in the blood approximately 1 hour post-dose across all doses studied (0.5 mg to 600 mg). Absorption is considered to be mostly complete (86.9%). The absolute oral bioavailability is 33.8%.
Effect of food
The remibrutinib AUC increased by 33% and Cmax decreased by 5%, respectively, with a high-fat meal compared to the fasted state following administration of remibrutinib. Remibrutinib may be taken with or without food (see section 4.2).
Distribution
Remibrutinib is readily distributed to blood cells with a blood-to-plasma ratio of 0.813. Plasma protein binding amounts to 95.4% with no concentration dependence. Based on pooled data from population pharmacokinetic (PopPK) analysis, the volume of distribution at steady state was 58 litres (central compartment) and 1 180 litres (peripheral compartment).
Biotransformation
Remibrutinib is metabolised primarily by CYP3A4, leading to the formation of 18 inactive metabolites, all in low amounts in circulation. Remibrutinib was the most abundant compound in blood (16.7%).
In vitro studies
In vitro CYP metabolism is predominantly driven by CYP3A4. In vitro data showed that remibrutinib is a P-gp substrate.
Elimination
Remibrutinib has a mean elimination half-life ranging between 1 and 2 hours at steady state. The mean apparent oral clearance at steady state (CLss/F), as determined by the PopPK analysis, is 160 litres/h. Following intravenous administration of 100 mg [14C]-remibrutinib, excretion of radioactivity (remibrutinib and metabolites) was approximately 72.9% of the administered dose in faeces and 27.1% in urine. Renal excretion of unchanged remibrutinib after oral administration was below 1% of the dose.
Linearity/non-linearity
The pharmacokinetics of remibrutinib at steady state are approximately linear in the total daily dose range of 10 to 200 mg.
Pharmacokinetic/pharmacodynamic relationship(s)
Clinical pharmacokinetic and pharmacodynamic (PK/PD) data estimated a BTK occupancy ≥96% in blood maintained throughout the entire day with remibrutinib 25 mg twice daily.
Special populations
PopPK analysis showed that there are no clinically relevant effects of age (18 to 80 years), sex (63.5% females and 36.5% males), race/ethnicity (59.3% Non-Asian, 8.8% Mainland Chinese, 12.2% Japanese, and 19.7% Other Asian) and body weight (39 to 162 kg; mean 74.8 kg) on the PK of remibrutinib.
Renal impairment
The effects of renal impairment on remibrutinib pharmacokinetics have not been evaluated in a dedicated clinical study. In a PopPK analysis, no clinically meaningful relationship was observed between renal function tests and remibrutinib pharmacokinetics. In the PopPK analysis, there were 19.3%, 2.2% and 0.1% of subjects with mild, moderate and severe renal impairment, respectively.
Hepatic impairment
The Cmax and AUC of remibrutinib at steady state increased 1.85-fold and 2.15-fold in subjects with mild hepatic impairment (Child-Pugh class A), 1.65-fold and 2.07-fold in subjects with moderate hepatic impairment (Child-Pugh class B), and 1.99-fold and 3.12-fold in subjects with severe hepatic impairment (Child-Pugh class C), respectively, relative to subjects with normal hepatic function following an oral dose of 25 mg remibrutinib twice daily. There was no change in protein binding of remibrutinib in subjects with hepatic impairment as compared to subjects with normal hepatic function (see section 4.2).
Paediatric population
No pharmacokinetic studies were performed with remibrutinib in patients under 18 years of age.
5.3. Preclinical safety data
Remibrutinib inhibited primary antibody responses in rodent pharmacology studies and increased rat tail bleeding time in haemostasis assessments. These observations, which occurred at pharmacologically and clinically relevant exposures, were considered related to effects of remibrutinib on specific B cell and platelet functions, respectively. The non-clinical data revealed no further special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenicity, and phototoxicity.
Reproductive toxicity
In embryo-foetal development (EFD) studies in pregnant rabbits, increased foetal external malformations (open/opaque eyes, small jaws, hyperflexion of forelimbs) and maternal toxicity (transiently reduced food consumption and adverse clinical signs) occurred at approximately 141 times the maximum recommended human dose (MRHD) of 25 mg twice daily with a no observed adverse effect level (NOAEL)-based safety margin of 23 times the MRHD of 25 mg twice daily based on AUC. The foetal findings were considered unlikely to be secondary to the maternal toxicity. No effect on EFD was observed in rats, with NOAEL-based safety margin of 126-fold in terms of steady-state AUC compared to human exposure at the MRHD.
In a pre- and postnatal development (PPND) study in rats, remibrutinib induced adverse effects affecting maternal animals (moribundity and clinical signs of toxicity, slightly longer gestation lengths) and offspring up to lactation day 1 (slightly higher mean number of stillborn, dead or missing pups and smaller mean litter size), with NOAEL-based safety margin for maternal animals and offspring of approximately 67 times the MRHD of 25 mg twice daily based on AUC. No adverse effects were noted for the surviving offspring developing into adulthood.
In a fertility study in rats, remibrutinib did not impact fertility in female or male rats up to the maximum achievable exposures of 79 and 15 times higher than MRHD of 25 mg twice daily based on AUC.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.