RoACTEMRA Concentrate for solution for infusion Ref.[8353] Active ingredients: Tocilizumab
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639, Grenzach-Wyhlen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, Interleukin inhibitors
ATC code: L04AC07
Mechanism of action
Tocilizumab binds specifically to both soluble and membrane-bound IL-6 receptors (sIL-6R and mIL-6R). Tocilizumab has been shown to inhibit sIL-6R and mIL-6R-mediated signalling. IL-6 is a pleiotropic pro-inflammatory cytokine produced by a variety of cell types including T- and B-cells, monocytes and fibroblasts. IL-6 is involved in diverse physiological processes such as T-cell activation, induction of immunoglobulin secretion, induction of hepatic acute phase protein synthesis and stimulation of haemopoiesis. IL-6 has been implicated in the pathogenesis of diseases including inflammatory diseases, osteoporosis and neoplasia.
RA Patients
Pharmacodynamic effects
In clinical studies with tocilizumab, rapid decreases in CRP, erythrocyte sedimentation rate (ESR), serum amyloid A (SAA) and fibrinogen were observed. Consistent with the effect on acute phase reactants, treatment with tocilizumab was associated with reduction in platelet count within the normal range. Increases in haemoglobin levels were observed, through tocilizumab decreasing the IL-6 driven effects on hepcidin production to increase iron availability. In tocilizumab-treated patients, decreases in the levels of CRP to within normal ranges were seen as early as week 2, with decreases maintained while on treatment.
In healthy subjects administered tocilizumab in doses from 2 to 28 mg/kg, absolute neutrophil counts decreased to their lowest 3 to 5 days following administration. Thereafter, neutrophils recovered towards baseline in a dose dependent manner. Rheumatoid arthritis patients demonstrated a similar pattern of absolute neutrophil counts following tocilizumab administration (see section 4.8).
Clinical efficacy and safety
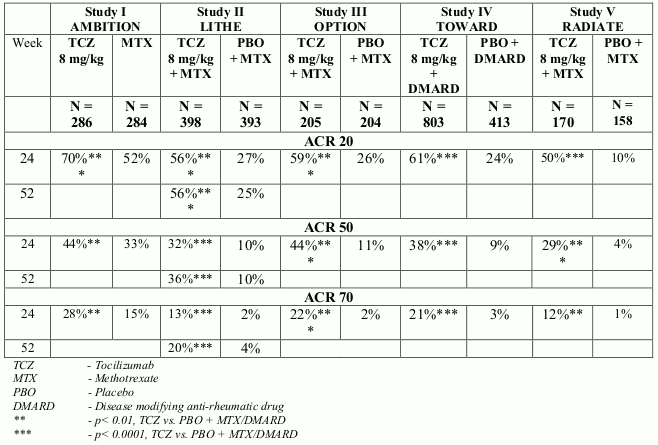
The efficacy of tocilizumab in alleviating the signs and symptoms of RA was assessed in five randomised, double-blind, multi-centre studies. Studies I-V enrolled patients ≥18 years of age with active RA diagnosed according to the American College of Rheumatology (ACR) criteria and who had at least eight tender and six swollen joints at baseline.
In Study I, tocilizumab was administered intravenously every four weeks as monotherapy. In Studies II, III and V, tocilizumab was administered intravenously every four weeks in combination with MTX vs. placebo and MTX. In Study IV, tocilizumab was administered intravenously every 4 weeks in combination with other DMARDs vs. placebo and other DMARDs. The primary endpoint for each of the five studies was the proportion of patients who achieved an ACR 20 response at week 24.
Study I evaluated 673 patients who had not been treated with MTX within six months prior to randomisation and who had not discontinued previous MTX treatment as a result of clinically important toxic effects or lack of response. The majority (67%) of patients were MTX-naïve. Doses of 8 mg/kg of tocilizumab were given every four weeks as monotherapy. The comparator group was weekly MTX (dose titrated from 7.5 mg to a maximum of 20 mg weekly over an eight week period).
Study II, a two year study with planned analyses at week 24, week 52 and week 104, evaluated 1,196 patients who had an inadequate clinical response to MTX. Doses of 4 or 8 mg/kg of tocilizumab or placebo were given every four weeks as blinded therapy for 52 weeks in combination with stable MTX (10 mg to 25 mg weekly). After week 52, all patients could receive open-label treatment with tocilizumab 8 mg/kg. Of the patients who completed the study who were originally randomised to placebo + MTX, 86% received open-label tocilizumab 8 mg/kg in year 2. The primary endpoint at week 24 was the proportion of patients who achieved an ACR 20 response. At week 52 and week 104 the co-primary endpoints were prevention of joint damage and improvement in physical function.
Study III evaluated 623 patients who had an inadequate clinical response to MTX. Doses of 4 or 8 mg/kg tocilizumab or placebo were given every four weeks, in combination with stable MTX (10 mg to 25 mg weekly).
Study IV evaluated 1,220 patients who had an inadequate response to their existing rheumatologic therapy, including one or more DMARDs. Doses of 8 mg/kg tocilizumab or placebo were given every four weeks in combination with stable DMARDs.
Study V evaluated 499 patients who had an inadequate clinical response or were intolerant to one or more TNF antagonist therapies. The TNF antagonist therapy was discontinued prior to randomisation. Doses of 4 or 8 mg/kg tocilizumab or placebo were given every four weeks in combination with stable MTX (10 mg to 25 mg weekly).
Clinical response
In all studies, patients treated with tocilizumab 8 mg/kg had statistically significant higher ACR 20, 50, 70 response rates at 6 months compared to control (Table 3). In study I, superiority of tocilizumab 8 mg/kg was demonstrated against the active comparator MTX.
The treatment effect was similar in patients independent of rheumatoid factor status, age, gender, race, number of prior treatments or disease status. Time to onset was rapid (as early as week 2) and the magnitude of response continued to improve with duration of treatment. Continued durable responses were seen for over 3 years in the open label extension studies I-V.
In patients treated with tocilizumab 8 mg/kg, significant improvements were noted on all individual components of the ACR response including: tender and swollen joint counts; patients and physician global assessment; disability index scores; pain assessment and CRP compared to patients receiving placebo plus MTX or other DMARDs in all studies.
Patients in studies I–V had a mean Disease Activity Score (DAS28) of 6.5–6.8 at baseline. Significant reduction in DAS28 from baseline (mean improvement) of 3.1–3.4 were observed in tocilizumab-treated patients compared to control patients (1.3-2.1). The proportion of patients achieving a DAS28 clinical remission (DAS28 <2.6) was significantly higher in patients receiving tocilizumab (28–34%) compared to 1–12% of control patients at 24 weeks. In study II, 65% of patients achieved a DAS28 <2.6 at week 104 compared to 48% at 52 weeks and 33% of patients at week 24.
In a pooled analysis of studies II, III and IV, the proportion of patients achieving an ACR 20, 50 and 70 response was significantly higher (59% vs. 50%, 37% vs. 27%, 18% vs. 11%, respectively) in the tocilizumab 8 mg/kg plus DMARD vs. the tocilizumab 4 mg/kg plus DMARD group (p<0.03). Similarly the proportion of patients achieving a DAS28 remission (DAS28 <2.6) was significantly higher (31% vs. 16% respectively) in patients receiving tocilizumab 8 mg/kg plus DMARD than in patients receiving tocilizumab 4 mg/kg plus DMARD (p<0.0001).
Table 3. ACR responses in placebo-/MTX-/DMARDs-controlled studies (% patients):
Major Clinical Response
After 2 years of treatment with tocilizumab plus MTX, 14% of patients achieved a major clinical response (maintenance of an ACR70 response for 24 weeks or more).
Radiographic response
In Study II, in patients with an inadequate response to MTX, inhibition of structural joint damage was assessed radiographically and expressed as change in modified Sharp score and its components, the erosion score and joint space narrowing score. Inhibition of joint structural damage was shown with significantly less radiographic progression in patients receiving tocilizumab compared to control (Table 4).
In the open-label extension of Study II the inhibition of progression of structural joint damage in tocilizumab plus MTX-treated patients was maintained in the second year of treatment. The mean change from baseline at week 104 in total Sharp-Genant score was significantly lower for patients randomised to tocilizumab 8 mg/kg plus MTX (p<0.0001) compared with patients who were randomised to placebo plus MTX.
Table 4. Radiographic mean changes over 52 weeks in Study II:
| PBO + MTX (+ TCZ from week 24) N=393 | TCZ 8 mg/kg + MTX N=398 | |
|---|---|---|
| Total Sharp-Genant score | 1.13 | 0.29* |
| Erosion score | 0.71 | 0.17* |
| JSN score | 0.42 | 0.12** |
PBO – Placebo
MTX – Methotrexate
TCZ – Tocilizumab
JSN – Joint space narrowing
* - p≤0.0001, TCZ vs. PBO + MTX
** - p<0.005, TCZ vs. PBO + MTX
Following 1 year of treatment with tocilizumab plus MTX, 85% of patients (n=348) had no progression of structural joint damage, as defined by a change in the Total Sharp Score of zero or less, compared with 67% of placebo plus MTX-treated patients (n=290) (p ≤ 0.001). This remained consistent following 2 years of treatment (83%; n=353). Ninety three percent (93%; n=271) of patients had no progression between week 52 and week 104.
Health-related and quality of life outcomes
Tocilizumab-treated patients reported an improvement in all patient-reported outcomes (Health Assessment Questionnaire Disability Index – HAQ-DI), Short Form-36 and Functional Assessment of Chronic Illness Therapy questionnaires. Statistically significant improvements in HAQ-DI scores were observed in patients treated with RoActemra compared with patients treated with DMARDs. During the open-label period of Study II, the improvement in physical function has been maintained for up to 2 years. At Week 52, the mean change in HAQ-DI was -0.58 in the tocilizumab 8 mg/kg plus MTX group compared with -0.39 in the placebo + MTX group. The mean change in HAQ-DI was maintained at Week 104 in the tocilizumab 8 mg/kg plus MTX group (-0.61).
Haemoglobin levels
Statistically significant improvements in haemoglobin levels were observed with tocilizumab compared with DMARDs (p<0.0001) at week 24. Mean haemoglobin levels increased by week 2 and remained within normal range through to week 24.
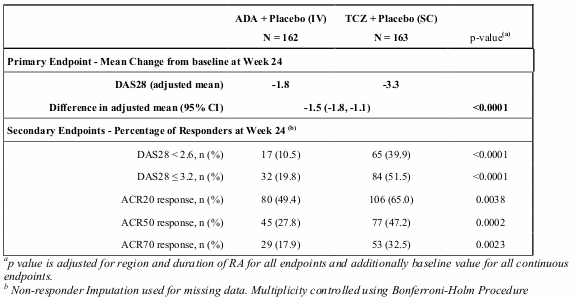
Tocilizumab versus adalimumab in monotherapy
Study VI (WA19924), a 24 week double-blinded study that compared tocilizumab monotherapy with adalimumab monotherapy, evaluated 326 patients with RA who were intolerant of MTX or where continued treatment with MTX was considered inappropriate (including MTX inadequate responders). Patients in the tocilizumab arm received an intravenous (IV) infusion of tocilizumab (8 mg/kg) every 4 weeks (q4w) and a subcutaneous (SC) placebo injection every 2 weeks (q2w). Patients in the adalimumab arm received an adalimumab SC injection (40 mg) q2w plus an IV placebo infusion q4w. A statistically significant superior treatment effect was seen in favour of tocilizumab over adalimumab in control of disease activity from baseline to week 24 for the primary endpoint of change in DAS28 and for all secondary endpoints (Table 5).
Table 5. Efficacy Results for Study VI (WA19924):
The overall clinical adverse event profile was similar between tocilizumab and adalimumab. The proportion of patients with serious adverse events was balanced between the treatment groups (tocilizumab 11.7% vs. adalimumab 9.9%). The types of adverse drug reactions in the tocilizumab arm were consistent with the known safety profile of tocilizumab and adverse drug reactions were reported at a similar frequency compared with Table 1. A higher incidence of infections and infestations was reported in the tocilizumab arm (48% vs. 42%), with no difference in the incidence of serious infections (3.1%). Both study treatments induced the same pattern of changes in laboratory safety parameters (decreases in neutrophil and platelet counts, increases in ALT, AST and lipids), however, the magnitude of change and the frequency of marked abnormalities was higher with tocilizumab compared with adalimumab. Four (2.5%) patients in the tocilizumab arm and two (1.2%) patients in the adalimumab arm experienced CTC grade 3 or 4 neutrophil count decreases. Eleven (6.8%) patients in the tocilizumab arm and five (3.1%) patients in the adalimumab arm experienced ALT increases of CTC grade 2 or higher. The mean LDL increase from baseline was 0.64 mmol/L (25 mg/dL) for patients in the tocilizumab arm and 0.19 mmol/L (7 mg/dL) for patients in the adalimumab arm. The safety observed in the tocilizumab arm was consistent with the known safety profile of tocilizumab and no new or unexpected adverse drug reactions were observed (see Table 1).
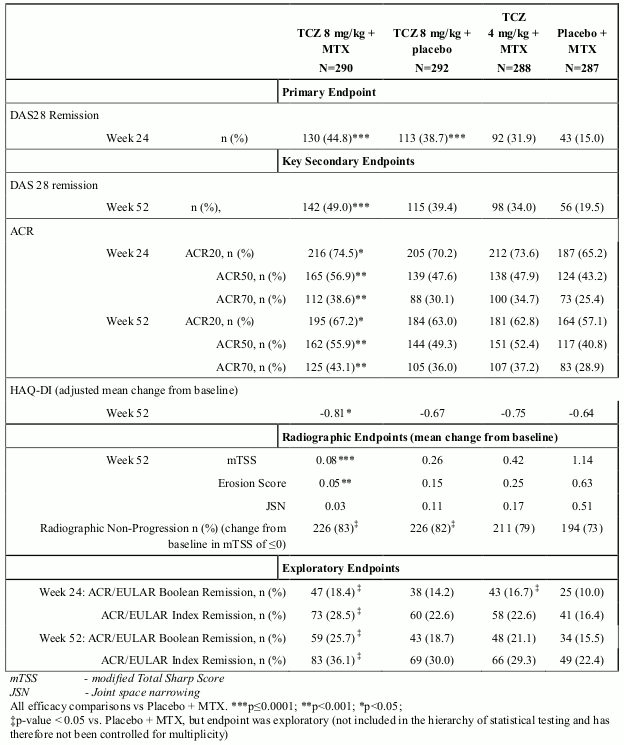
MTX naïve, Early RA
Study VII (WA19926), a 2 year study with the planned primary analysis at week 52 evaluated 1162 MTX-naïve adult patients with moderate to severe, active early RA (mean disease duration ≤6 months). Approximately 20% of patients had received prior treatment with DMARDs other than MTX. This study evaluated the efficacy of IV tocilizumab 4 or 8 mg/kg every 4 weeks/MTX combination therapy, IV tocilizumab 8 mg/kg monotherapy and MTX monotherapy in reducing the signs and symptoms and rate of progression of joint damage for 104 weeks. The primary endpoint was the proportion of patients achieving DAS28 remission (DAS28 <2.6) at week 24. A significantly higher proportion of patients in the tocilizumab 8 mg/kg + MTX and tocilizumab monotherapy groups met the primary endpoint compared with MTX alone. The tocilizumab 8 mg/kg + MTX group also showed statistically significant results across the key secondary endpoints. Numerically greater responses compared with MTX alone were observed in the tocilizumab 8 mg/kg monotherapy group in all secondary endpoints, including radiographic endpoints. In this study, ACR/EULAR remission (Boolean and Index) were also analysed as pre-specified exploratory endpoints, with higher responses observed in the tocilizumab groups. The results from study VII are shown in Table 6.
Table 6. Efficacy Results for Study VII (WA19926) on MTX-naïve, early RA patients:
Paediatric population
sJIA Patients
Clinical efficacy
The efficacy of tocilizumab for the treatment of active sJIA was assessed in a 12 week randomised, double blind, placebo-controlled, parallel group, two arm study. Patients included in the trial had a total disease duration of at least 6 months and active disease but were not experiencing an acute flare requiring corticosteroid doses of more than 0.5 mg/kg prednisone equivalent. Efficacy for the treatment of macrophage activation syndrome has not been investigated.
Patients (treated with or without MTX) were randomised (tocilizumab:placebo = 2:1) to one of two treatment groups, 75 patients received tocilizumab infusions every two weeks, either 8 mg/kg for patients ≥30 kg or 12 mg/kg for patients <30 kg and 37 patients were assigned to receiving placebo infusions every two weeks. Corticosteroid tapering was permitted from week six for patients who achieved a JIA ACR70 response. After 12 weeks or at the time of escape, due to disease worsening, patients were treated in the open label phase at weight appropriate dosing.
Clinical response
The primary endpoint was the proportion of patients with at least 30% improvement in the JIA ACR core set (JIA ACR30 response) at week 12 and absence of fever (no temperature recording ≥ 37.5°C in the preceding 7 days). Eighty five percent (64/75) of tocilizumab treated patients and 24.3% (9/37) of placebo treated patients achieved this endpoint. These proportions were highly significantly different (p<0.0001).
The percent of patients achieving JIA ACR 30, 50, 70 and 90 responses are shown in Table 7.
Table 7. JIA ACR response rates at week 12 (% patients):
| Response Rate | Tocilizumab N=75 | Placebo N=37 |
|---|---|---|
| JIA ACR 30 | 90.7%1 | 24.3% |
| JIA ACR 50 | 85.3%1 | 10.8% |
| JIA ACR 70 | 70.7%1 | 8.1% |
| JIA ACR 90 | 37.3%1 | 5.4% |
1 p<0.0001, tocilizumab vs. placebo
Systemic Effects
In the tocilizumab treated patients, 85% who had fever due to sJIA at baseline were free of fever (no temperature recording ≥37.5°C in the preceding 14 days) at week 12 versus 21% of placebo patients (p<0.0001).
The adjusted mean change in the pain VAS after 12 weeks of tocilizumab treatment was a reduction of 41 points on a scale of 0-100 compared to a reduction of 1 for placebo patients (p<0.0001).
Corticosteroid Tapering
Patients achieving a JIA ACR70 response were permitted corticosteroid dose reduction. Seventeen (24%) tocilizumab treated patients versus 1 (3%) placebo patient were able to reduce their dose of corticosteroid by at least 20% without experiencing a subsequent JIA ACR30 flare or occurrence of systemic symptoms to week 12 (p=0.028). Reductions in corticosteroids continued, with 44 patients off oral corticosteroids at week 44, while maintaining JIA ACR responses.
Health related and quality of life outcomes
At week 12, the proportion of tocilizumab treated patients showing a minimally clinically important improvement in the Childhood Health Assessment Questionnaire – Disability Index (defined as an individual total score decrease of ≥0.13) was significantly higher than in placebo treated patients, 77% versus 19% (p<0.0001).
Laboratory Parameters
Fifty out of seventy five (67%) tocilizumab treated patients had a haemoglobin <LLN at baseline. Forty (80%) of these patients had an increase in their haemoglobin to within the normal range at week 12, in comparison to 2 out of 29 (7%) of placebo treated patients with haemoglobin <LLN at baseline (p<0.0001).
pJIA Patients
Clinical efficacy
The efficacy of tocilizumab was assessed in a three-part study WA19977 including an open-label extension in children with active pJIA. Part I consisted of a 16-week active tocilizumab treatment lead-in period (n=188) followed by Part II, a 24-week randomized double-blind placebo-controlled withdrawal period (n=163), followed by Part III, a 64-week open-label period. In Part 1, eligible patients ≥30 kg received tocilizumab at 8 mg/kg IV every 4 weeks for 4 doses. Patients <30 kg were randomized 1:1 to receive either tocilizumab 8 mg/kg or 10 mg/kg IV every 4 weeks for 4 doses. Patients who completed Part I of the study and achieved at least a JIA ACR30 response at week 16 compared to baseline were eligible to enter the blinded withdrawal period (Part II) of the study. In Part II, patients were randomized to tocilizumab (same dose received in Part I) or placebo in a 1:1 ratio, stratified by concurrent MTX use and concurrent corticosteroid use. Each patient continued in Part II of the study until Week 40 or until the patient satisfied JIA ACR30 flare criteria (relative to Week 16) and qualified for escape to tocilizumab therapy (same dose received in Part I).
Clinical response
The primary endpoint was the proportion of patients with a JIA ACR30 flare at week 40 relative to week 16. Forty eight percent (48.1%, 39/81) of the patients treated with placebo flared compared with 25.6% (21/82) of tocilizumab treated patients. These proportions were statistically significantly different (p=0.0024).
At the conclusion of Part I, JIA ACR 30/50/70/90 responses were 89.4%, 83.0%, 62.2%, and 26.1%, respectively.
During the withdrawal phase (Part II), the percentage of patients achieving JIA ACR 30, 50, and 70 responses at Week 40 relative to baseline are shown in Table 8. In this statistical analysis, patients who flared (and escaped to TCZ) during Part II or who withdrew, were classified as non-responders. An additional analyses of JIA ACR responses, considering observed data at Week 40, regardless of flare status, showed that by Week 40, 95.1% of patients who had received continuous TCZ therapy, had achieved JIA ACR30 or higher.
Table 8. JIA ACR Response Rates at Week 40 Relative to baseline (Percentage of Patients):
| Response Rate | Tocilizumab N=82 | Placebo N=81 |
|---|---|---|
| ACR 30 | 74.4%* | 54.3%* |
| ACR 50 | 73.2%* | 51.9%* |
| ACR 70 | 64.6%* | 42.0%* |
* p<0.01, tocilizumab vs. placebo
The number of active joints was significantly reduced compared to baseline in patients receiving tocilizumab compared to placebo (adjusted mean changes of -14.3 vs -11.4, p=0.0435). The physician’s global assessment of disease activity, as measured on a 0-100 mm scale, showed a greater reduction in disease activity for tocilizumab compared to placebo (adjusted mean changes of -45.2 mm vs -35.2 mm, p=0.0031).
The adjusted mean change in the pain VAS after 40 weeks of tocilizumab treatment was 32.4 mm on a 0-100 mm scale compared to a reduction of 22.3 mm for placebo patients (highly statistically significant; p=0.0076).
The ACR response rates were numerically lower for patients with prior biologic treatment as shown in Table 9 below.
Table 9. Number and Proportion of Patients with a JIA ACR30 Flare and Proportion of Patients with JIA ACR30/50/70/90 Responses at Week 40, by Previous Biologic Use (ITT Population – Study Part II):
| Placebo | All TCZ | |||
|---|---|---|---|---|
| Biologic Use | Yes (N=23) | No (N=58) | Yes (N=27) | No (N=55) |
| JIA ACR30 Flare 18 | (78.3) | 21 (36.2) | 12 (44.4) | 9 (16.4) |
| JIA ACR30 Response 6 | (26.1) | 38 (65.5) | 15 (55.6) | 46 (83.6) |
| JIA ACR50 Response 5 | (21.7) | 37 (63.8) | 14 (51.9) | 46 (83.6) |
| JIA ACR70 Response 2 | (8.7) | 32 (55.2) | 13 (48.1) | 40 (72.7) |
| JIA ACR90 Response 2 | (8.7) | 17 (29.3) | 5 (18.5) | 32 (58.2) |
Patients randomized to tocilizumab had fewer ACR30 flares and higher overall ACR responses than patients receiving placebo regardless of a history of prior biologic use.
CRS
The efficacy of RoActemra for the treatment of CRS was assessed in a retrospective analysis of data from clinical trials of CAR T-cell therapies (tisagenlecleucel and axicabtagene ciloleucel) for hematological malignancies. Evaluable patients had been treated with tocilizumab 8 mg/kg (12 mg/kg for patients <30 kg) with or without additional high-dose corticosteroids for severe or life-threatening CRS; only the first episode of CRS was included in the analysis. The efficacy population for the tisagenlecleucel cohort included 28 males and 23 females (total 51 patients) of median age 17 years (range, 3–68 years). The median time from start of CRS to first dose of tocilizumab was 3 days (range, 0–18 days). Resolution of CRS was defined as lack of fever and off vasopressors for at least 24 hours. Patients were considered responders if CRS resolved within 14 days of the first dose of tocilizumab, if no more than 2 doses of RoActemra were needed, and no drugs other than RoActemra and corticosteroids were used for treatment. Thirty-nine patients (76.5%; 95% CI: 62.5%–87.2%) achieved a response. In an independent cohort of 15 patients (range: 9–75 years old) with axicabtagene ciloleucel-induced CRS, 53% responded.
The European Medicines Agency has waived the obligation to submit the results of studies with RoActemra in all subsets of the paediatric population in treatment of cytokine release syndrome associated with chimeric antigen receptor (CAR) T cell therapy.
Pharmacokinetic properties
RA Patients
Intravenous use
The pharmacokinetics of tocilizumab were determined using a population pharmacokinetic analysis on a database composed of 3552 RA patients treated with a one-hour infusion of 4 or 8 mg/kg tocilizumab every 4 weeks for 24 weeks or with 162 mg tocilizumab given subcutaneously either once a week or every other week for 24 weeks.
The following parameters (predicted mean ± SD) were estimated for a dose of 8 mg/kg tocilizumab given every 4 weeks: steady-state area under curve (AUC) = 38000 ± 13000 h μg/mL, trough concentration (Cmin) = 15.9 ± 13.1 μg/mL and maximum concentration (Cmax) = 182 ± 50.4 μg/mL, and the accumulation ratios for AUC and Cmax were small, 1.32 and 1.09, respectively. The accumulation ratio was higher for Cmin (2.49), which was expected based on the non-linear clearance contribution at lower concentrations. Steady-state was reached following the first administration for Cmax and after 8 and 20 weeks for AUC and Cmin, respectively. Tocilizumab AUC, Cmin and Cmax increased with increase of body weight. At body weight ≥100 kg, the predicted mean (± SD) steady-state AUC, Cmin and Cmax of tocilizumab were 50000 ± 16800 μg•h/mL, 24.4 ± 17.5 μg/mL, and 226 ± 50.3 μg/mL, respectively, which are higher than mean exposure values for the patient population (i.e. all body weights) reported above. The dose-response curve for tocilizumab flattens at higher exposure, resulting in smaller efficacy gains for each incremental increase in tocilizumab concentration such that clinically meaningful increases in efficacy were not demonstrated in patients treated with >800 mg of tocilizumab. Therefore, tocilizumab doses exceeding 800 mg per infusion are not recommended (see section 4.2).
Distribution
In RA patients the central volume of distribution was 3.72, the peripheral volume of distribution was 3.35 resulting in a volume of distribution at steady state of 7.07.
Elimination
Following intravenous administration, tocilizumab undergoes biphasic elimination from the circulation. The total clearance of tocilizumab was concentration-dependent and is the sum of the linear and non-linear clearance. The linear clearance was estimated as a parameter in the population pharmacokinetic analysis and was 9.5 mL/h. The concentration-dependent non-linear clearance plays a major role at low tocilizumab concentrations. Once the non-linear clearance pathway is saturated, at higher tocilizumab concentrations, clearance is mainly determined by the linear clearance.
The t1/2 of tocilizumab was concentration-dependent. At steady-state following a dose of 8 mg/kg every 4 weeks, the effective t½ decreased with decreasing concentrations within a dosing interval from 18 days to 6 days.
Linearity
Pharmacokinetic parameters of tocilizumab did not change with time. A more than dose-proportional increase in the AUC and Cmin was observed for doses of 4 and 8 mg/kg every 4 weeks. Cmax increased dose-proportionally. At steady-state, predicted AUC and Cmin were 3.2 and 30 fold higher at 8 mg/kg as compared to 4 mg/kg, respectively.
Special populations
Renal impairment: No formal study of the effect of renal impairment on the pharmacokinetics of tocilizumab has been conducted. Most of the patients in the population pharmacokinetic analysis had normal renal function or mild renal impairment. Mild renal impairment (creatinine clearance based on Cockcroft-Gault <80 mL/min and ≥50 mL/min) did not impact the pharmacokinetics of tocilizumab.
Hepatic impairment: No formal study of the effect of hepatic impairment on the pharmacokinetics of tocilizumab has been conducted.
Age, gender and ethnicity: Population pharmacokinetic analyses in RA patients, showed that age, gender and ethnic origin did not affect the pharmacokinetics of tocilizumab.
sJIA Patients
The pharmacokinetics of tocilizumab were determined using a population pharmacokinetic analysis on a database composed of 140 sJIA patients treated with 8 mg/kg IV every 2 weeks (patients with a body weight ≥30 kg ) 12 mg/kg IV every 2 weeks (patients with a body weight <30 kg), 162 mg SC every week (patients weighing ≥30 kg), 162 mg SC every 10 days or every 2 weeks (patients weighing below 30 kg).
Table 10. Predicted mean ± SD PK parameters at steady-state after IV dosing in sJIA:
| RoActemra PK Parameter | 8 mg/kg Q2W≥30 kg | 12 mg/kg Q2W below 30 kg |
|---|---|---|
| Cmax (µg/mL) | 256 ± 60.8 | 274 ± 63.8 |
| Ctrough (µg/mL) | 69.7 ± 29.1 | 68.4 ± 30.0 |
| Cmean (µg/mL) | 119 ± 36.0 | 123 ± 36.0 |
| Accumulation Cmax | 1.42 | 1.37 |
| Accumulation Ctrough | 3.20 | 3.41 |
| Accumulation Cmean or AUCτ* | 2.01 | 1.95 |
* τ = 2 weeks for IV regimens
After IV dosing, approximately 90% of the steady-state was reached by week 8 for both the 12 mg/kg (BW<30 kg) and 8 mg/kg Q2W (BW≥30 kg) regimens.
In sJIA patients, the central volume of distribution was 1.87 L and the peripheral volume of distribution was 2.14 L resulting in a volume of distribution at a steady state of 4.01 L. The linear clearance estimated as a parameter in the population pharmacokinetic analysis, was 5.7 mL/h.
The half life of tocilizumab in sJIA patients is up to 16 days for the two body weight categories (8 mg/kg for body weight ≥30 kg or 12 mg/kg for body weight <30 kg) at week 12.
pJIA Patients
The pharmacokinetics of tocilizumab in pJIA patients was characterized by a population pharmacokinetic analysis which included 237 patients who were treated with 8 mg/kg IV every 4 weeks (patients weighing ≥30 kg), 10 mg/kg IV every 4 weeks (patients weighing below 30 kg), 162 mg SC every 2 weeks (patients weighing ≥30 kg), or 162 mg SC every 3 weeks (patients weighing below 30 kg).
Table 11. Predicted mean ± SD PK parameters at steady-state after IV dosing in pJIA:
| RoActemra PK Parameter | 8 mg/kg Q4W≥30 kg | 10 mg/kg Q4W below 30 kg |
|---|---|---|
| Cmax (µg/mL) | 183 ± 42.3 | 168 ± 24.8 |
| Ctrough (µg/mL) | 6.55 ± 7.93 | 1.47 ± 2.44 |
| Cmean (µg/mL) | 42.2 ± 13.4 | 31.6 ± 7.84 |
| Accumulation Cmax | 1.04 | 1.01 |
| Accumulation Ctrough | 2.22 | 1.43 |
| Accumulation Cmean or AUCτ* | 1.16 | 1.05 |
* τ = 4 weeks for IV regimens
After IV dosing, approximately 90% of the steady-state was reached by week 12 for the 10 mg/kg (BW<30 kg), and by week 16 for the 8 mg/kg (BW≥30 kg) dose.
The half life of tocilizumab in pJIA patients is up to 16 days for the two body weight categories (8 mg/kg for body weight ≥30 kg or 10 mg/kg for body weight <30 kg) during a dosing interval at steady state.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity and genotoxicity.
Carcinogenicity studies were not performed because IgG1 monoclonal antibodies are not deemed to have intrinsic carcinogenic potential. Available non-clinical data demonstrated the effect of IL-6 on malignant progression and apoptosis resistance to various cancer types. This data does not suggest a relevant risk for cancer initiation and progression under tocilizumab treatment. Additionally, proliferative lesions were not observed in a 6-month chronic toxicity study in cynomolgus monkeys or in IL-6 deficient mice.
Available non-clinical data do not suggest an effect on fertility under tocilizumab treatment. Effects on endocrine active and reproductive system organs were not observed in a chronic cynomolgus monkey toxicity study and reproductive performance was not affected in IL-6 deficient mice. Tocilizumab administered to cynomolgus monkeys during early gestation, was observed to have no direct or indirect harmful effect on pregnancy or embryonal-foetal development. However, a slight increase in abortion/embryonal-foetal death was observed with high systemic exposure (>100 x human exposure) in the 50 mg/kg/day high-dose group compared to placebo and other low-dose groups. Although IL-6 does not seem to be a critical cytokine for foetal growth or the immunological control of the maternal/foetal interface, a relation of this finding to tocilizumab cannot be excluded.
Treatment with a murine analogue did not exert toxicity in juvenile mice. In particular, there was no impairment of skeletal growth, immune function and sexual maturation.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.