Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Sanofi-aventis groupe, 54, rue La Boétie, 75008 Paris, France
Pharmacotherapeutic group: Drugs used in diabetes, insulins and analogues for injection, long-acting.
ATC Code: A10AE54.
Suliqua combines two active substances with complementary mechanisms of action to improve glycaemic control: insulin glargine, a basal insulin analogue (mainly targeting fasting plasma glucose), and lixisenatide, a GLP-1 receptor agonist (mainly targeting postprandial glucose).
The primary activity of insulin, including insulin glargine, is regulation of glucose metabolism. Insulin and its analogues lower blood glucose by stimulating peripheral glucose uptake, especially by skeletal muscle and fat, and by inhibiting hepatic glucose production. Insulin inhibits lipolysis and proteolysis, and enhances protein synthesis.
Lixisenatide is a GLP-1 receptor agonist. The GLP-1 receptor is the target for native GLP-1, an endogenous incretin hormone that potentiates glucose-dependent insulin secretion from beta cells and suppresses glucagon from alpha cells in the pancreas.
Lixisenatide stimulates insulin secretion when blood glucose is increased but not at normoglycaemia, which limits the risk of hypoglycaemia. In parallel, glucagon secretion is suppressed. In case of hypoglycaemia, the rescue mechanism of glucagon secretion is preserved. A postprandial injection of Lixisenatide also slows gastric emptying thereby reducing the rate at which meal-derived glucose is absorbed and appears in the circulation.
The combination of insulin glargine and lixisenatide has no impact on the pharmacodynamics of insulin glargine. The impact of the combination of insulin glargine and lixisenatide on the pharmacodynamics of lixisenatide has not been studied in phase 1 studies. Consistent with a relatively constant concentration/time profile of insulin glargine over 24 hours with no pronounced peak when administered alone, the glucose utilisation rate/time profile was similar when given in the insulin glargine/lixisenatide combination. The time course of action of insulins, including Suliqua, may vary between individuals and within the same individual.
In clinical studies with insulin glargine (100 units/ml) the glucose-lowering effect on a molar basis (i.e., when given at the same doses) of intravenous insulin glargine is approximately the same as that for human insulin.
In a 28-day placebo-controlled study in patients with type 2 diabetes 5 to 20 mcg lixisenatide resulted in a statistically significant decreases in postprandial blood glucose after breakfast, lunch and dinner.
Following a standardised labelled test meal, in the study referred to above, it was confirmed that lixisenatide slows gastric emptying, thereby reducing the rate of postprandial glucose absorption. The slowing effect of gastric emptying was maintained at the end of the study.
The safety and effectiveness of Suliqua on glycaemic control were evaluated in three randomised clinical studies in patients with type 2 diabetes mellitus:
In each of the active-controlled clinical studies, treatment with Suliqua produced clinically and statistically significant improvements in haemoglobin A1c (HbA1c). Reaching lower HbA1c levels and achieving greater HbA1c reduction did not increase rates of hypoglycaemia with combination treatment versus insulin glargine alone (see section 4.8).
In the Add-on to metformin clinical study the treatment was started at 10 dose steps (10 units insulin glargine and 5 mcg lixisenatide). In the switch from basal insulin clinical study the starting dose was 20 dose steps (20 units insulin glargine and 10 mcg lixisenatide) or 30 dose steps, (30 units insulin glargine and 10 mcg lixisenatide), see section 4.2, depending on the previous insulin dose. In both studies the dose was titrated once weekly, based on fasting self-measured plasma glucose values.
A total of 1170 patients with type 2 diabetes were randomised in an open label, 30-week, activecontrolled study to evaluate the efficacy and safety of Suliqua compared to the individual components, insulin glargine (100 units/ml) and lixisenatide (20 mcg).
Patients with type 2 diabetes, treated with metformin alone or metformin and a second OAD treatment that could be a sulfonylurea or a glinide or a SGLT-2 inhibitor or a dipeptidyl peptidase-4 (DPP-4) inhibitor, and who were not adequately controlled with this treatment (HbA1c range 7.5% to 10% for patients previously treated with metformin alone and 7% to 9% for patients previously treated with metformin and a second oral anti-diabetic treatment) entered a run-in period for 4 weeks. During this run-in phase metformin treatment was optimised and any other OADs were discontinued. At the end of the run-in period, patients who remained inadequately controlled (HbA1c between 7% and 10%) were randomised to either Suliqua, insulin glargine or lixisenatide. Of the 1479 patients who started the run-in phase, 1170 were randomised. The main reasons for not entering the randomised phase were FPG value >13.9 mmol/L and HbA1c value <7% or >10% at the end of the run-in phase
The randomised type 2 diabetes population had the following characteristics: Mean age was 58.4 years with the majority (57.1%) being aged of 50 to 64 years, and 50.6 percent were male. The mean BMI at baseline was 31.7 kg/m² with 63.4% of patients having a BMI ≥30 kg/m². The mean duration of diabetes was approximately 9 years. Metformin was a mandatory background therapy and 58% of patients received a second OAD at screening, being a sulfonylurea in 54% of patients.
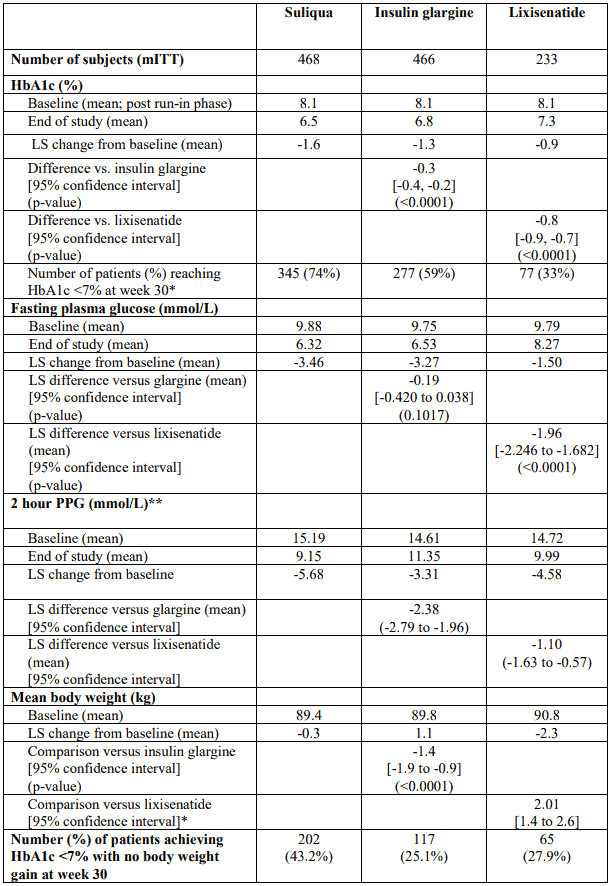
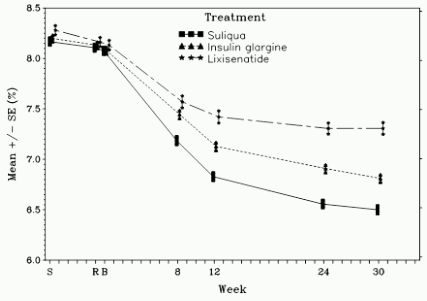
At week 30, Suliqua provided statistically significant improvement in HbA1c (p-value <0.0001) compared to the individual components. In a pre-specified analysis of this primary endpoint, the differences observed were consistent with regard to baseline HbA1c (<8% or ≥8%) or baseline OAD use (metformin alone or metformin plus second OAD).
See table and figure below for the other endpoints in the study.
Table 3. Results at 30 weeks – Add-on to metformin clinical study (mITT population):
Figure 1. Mean HbA1c (%) by visit during 30-week randomised treatment period – mITT population:
Patients in the Suliqua group reported a statistically significantly greater decrease in the average 7-point self-monitored plasma glucose (SMPG) profile from baseline to Week 30 (-3.35 mmol/L) compared to patients in the insulin glargine group (-2.66 mmol/L; difference -0.69 mmol/L) and patients in the lixisenatide group (-1.95 mmol/L; difference -1.40 mmol/L) (p<0.0001 for both comparisons). At all time points, 30-week mean plasma glucose values were lower in the Suliqua group than in both the insulin glargine group and the lixisenatide group, with the only exception of the pre-breakfast value which was similar between the Suliqua group and the insulin glargine group.
A total of 736 patients with type 2 diabetes participated in a randomised, 30-week, active-controlled, open-label, 2-treatment arm, parallel-group, multicentre study to evaluate the efficacy and safety of Suliqua compared to insulin glargine (100 units/ml).
Patients screened had type 2 diabetes were treated with basal insulin for at least 6 months, receiving a stable daily dose of between 15 and 40 U alone or combined with 1 or 2 OADs (metformin or a sulfonylurea or a glinide or a SGLT-2 inhibitor or a DPP-4 inhibitor), had an HbA1c between 7.5% and 10% (mean HbA1c of 8.5% at screening) and a FPG less than or equal to 10.0 mmol/L or 11.1 mmol/L depending on their previous anti-diabetic treatment.
After screening, eligible patients (n=1018) entered a 6 week run-in phase where patients remained on or switched to insulin glargine, in case they took another basal insulin, and had their insulin dose titrated/stabilised while continuing metformin (if previously taken). Any other OADs were discontinued. At the end of the run-in period, patients with an HbA1c between 7 and 10%, FPG ≤7.77 mmol/L and insulin glargine daily dose of 20 to 50 units, were randomised to either Suliqua (n=367) or insulin glargine (n=369).
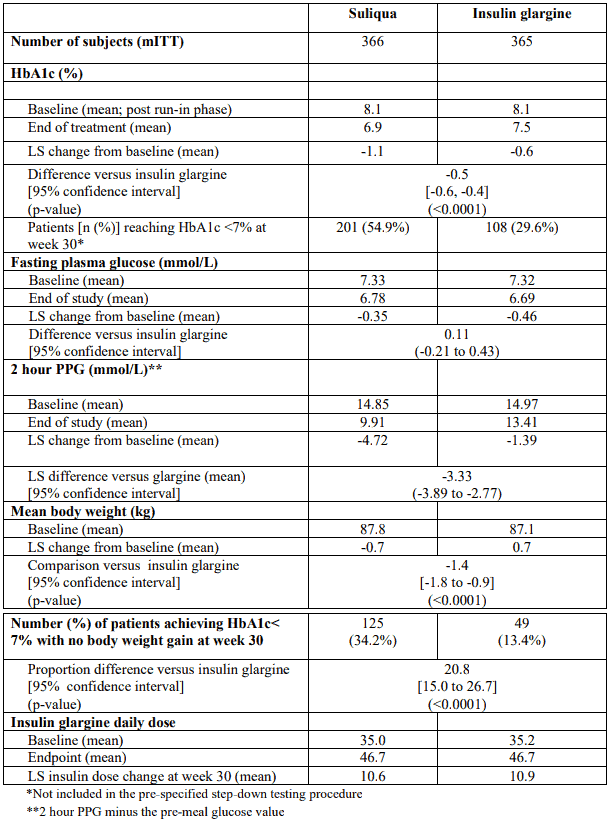
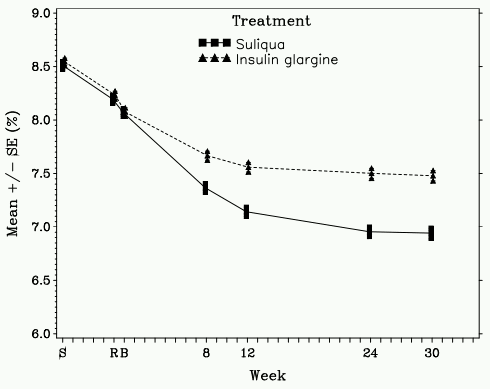
This type 2 diabetes population had the following characteristics: mean age was 60.0 years with the majority (56.3%) being aged of 50 to 64 years, and 53.3 percent were female. The mean BMI at baseline was 31.1 kg/m² with 57.3% of patients having a BMI ≥30 kg/m². The mean diabetes duration was approximately 12 years and the mean duration of previous basal insulin treatment was approximately 3 years. At screening 64.4% of patients were receiving insulin glargine as basal insulin and 95% received at least 1 concomitant OAD. At week 30, Suliqua provided statistically significant improvement in HbA1c (p-value <0.0001) compared to insulin glargine. See table and figure below for the other endpoints in the study.
Table 4. Results at 30 weeks – Study Type 2 diabetes uncontrolled on basal insulin mITT population:
Figure 2. Mean HbA1c (%) by visit during 30-week randomised treatment period – mITT population:
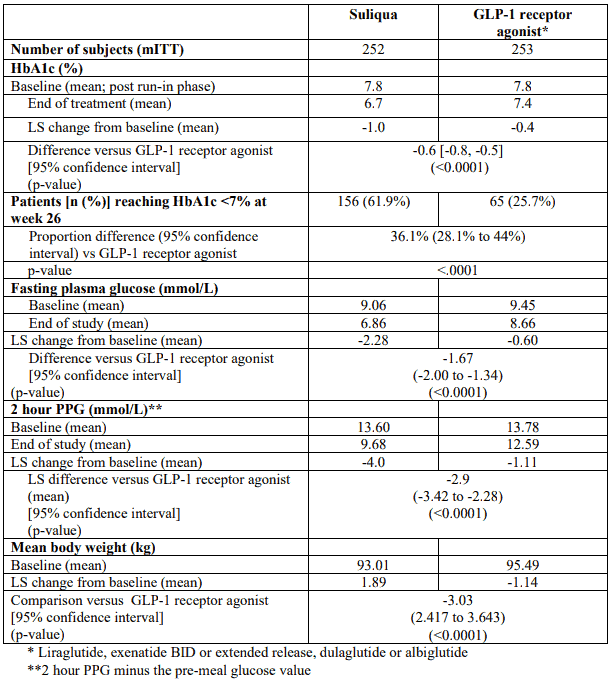
The efficacy and safety of Suliqua compared to unchanged pre-trial GLP-1 receptor agonist treatment were studied in a 26-week, randomised, open-label trial. The trial included 514 patients with type 2 diabetes mellitus inadequately controlled (HbA1c level of 7% to 9% both inclusive) while treated for at least 4 months with liraglutide or exenatide or for at least 6 months with dulaglutide, albiglutide or exenatide extended release, all at maximal tolerated dose, and metformin alone or in combination with pioglitazone, a SGLT-2 inhibitor or both. Eligible patients were randomised to either receive Suliqua or to continue their previous GLP-1 receptor agonist both on top of their previous oral anti-diabetic treatment.
At screening, 59.7% of the subjects received a once or twice-daily GLP-1 receptor agonist and 40.3% received a once weekly GLP-1 receptor agonist. At screening, 6.6% of the subjects received pioglitazone, and 10.1% a SGLT-2 inhibitor in combination with metformin. The study population had the following characteristics: mean age was 59.6 years, 52.5% of the subjects were male. The mean duration of diabetes was 11 years, the mean duration of previous GLP-1 receptor agonist treatment was 1.9 years, the mean BMI was approximately 32.9 kg/m², mean eGFR was 87.3 ml/min/1.73 m² and 90.7% of patients had an eGFR ≥60 ml/min.
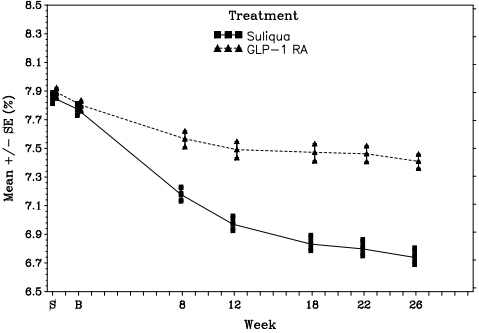
At week 26, Suliqua provided statistically significant improvement in HbA1c (p <0.0001). A prespecified analysis by GLP-1 receptor agonist subtype (once/twice daily or weekly formulation) used at screening showed that HbA1c change at week 26 was similar for each subgroup and consistent with the primary analysis for the overall population. The mean daily dose of Suliqua at Week 26 was 43.5 dose steps.
See table and figure below for the other endpoints in the study.
Table 5. Results at 26 weeks – Study Type 2 diabetes uncontrolled on GLP-1 receptor agonist mITT population:
Figure 3. Mean HbA1c (%) by visit during 26-week randomised treatment period- mITT population:
The concomitant use of Suliqua with SGLT2i is supported by subgroup analyses from three phase 3 randomised clinical studies (119 patients on the insulin glargine/lixisenatide fixed ratio combination (FRC) who also received SGLT2i).
One study conducted in Europe and North America included data from 26 patients (10.1%) who concomitantly received insulin glargine/lixisenatide FRC, metformin and an SGLT2i. Two more phase 3 studies from the dedicated Japanese clinical development program performed in patients not reaching sufficient glycaemic control on OADs provided data for 59 patients (22.7%) and 34 patients (21.1%), respectively, who concomitantly received SGLT2i and insulin glargine/lixisenatide FRC.
The data from these 3 studies show that initiation of Suliqua in patients inadequately controlled with a treatment including SGLT2i leads to improved change in HbA1c versus the comparators (insulin glargine, lixisenatide, liraglutide, exenatide BID or extended release, dulaglutide or albiglutide). There was no increased risk of hypoglycaemia and no relevant differences in the overall safety profile in SGLT2i users compared to non-users.
The cardiovascular safety of insulin glargine and lixisenatide has been established in the ORIGIN and ELIXA clinical studies, respectively. No dedicated cardiovascular outcome trial has been conducted with Suliqua.
The Outcome Reduction with Initial Glargine Intervention trial (i.e., ORIGIN) was an open-label, randomised, 12,537 patient study that compared insulin glargine 100 Units to standard care on the time to first occurrence of a major adverse cardiovascular event (MACE). MACE was defined as the composite of cardiovascular (CV) death, nonfatal myocardial infarction and nonfatal stroke. The median duration of study follow-up was 6.2 years. The incidence of MACE was similar between insulin glargine 100 Units and standard care in ORIGIN [Hazard Ratio (95% CI) for MACE; 1.02 (0.94, 1.11)].
The ELIXA study was a randomised, double-blind, placebo-controlled, multinational study that evaluated CV outcomes during treatment with lixisenatide in patients (n=6068) with type 2 diabetes mellitus after a recent Acute Coronary Syndrome. The primary composite efficacy endpoint was the time to the first occurrence of any of the following events: CV death, non-fatal myocardial infarction, non-fatal stroke, or hospitalisation for unstable angina. The median duration of study follow-up was 25.8 and 25.7 months in the lixisenatide group and the placebo group, respectively.
The incidence of the primary endpoint was similar in the lixisenatide (13.4%) and placebo (13.2%) groups: the hazard ratio (HR) for lixisenatide versus placebo was 1.017, with an associated 2-sided 95% confidence interval (CI) of 0.886 to 1.168.
The European Medicines Agency has waived the obligation to submit the results of studies with Suliqua in all subsets of the paediatric population in the treatment of type 2 diabetes mellitus (see section 4.2 for information on paediatric use).
The insulin glargine/lixisenatide ratio has no relevant impact on the PK of insulin glargine and lixisenatide in Suliqua.
After subcutaneous administration of insulin glargine/lixisenatide combinations to patients with type 1 diabetes, insulin glargine showed no pronounced peak. Exposure to insulin glargine following administration of the insulin glargine/lixisenatide combination was 86-88% compared to administration of separate simultaneous injections of insulin glargine and lixisenatide. This difference is not considered clinically relevant.
After subcutaneous administration of insulin glargine/lixisenatide combinations to patients with type 1 diabetes, the median tmax of lixisenatide was in the range of 2.5 to 3.0 hours. AUC was comparable while there was a small decrease in Cmax of lixisenatide of 22-34% compared with separate simultaneous administration of insulin glargine and lixisenatide, which is not likely to be clinically significant.
There are no clinically relevant differences in the rate of absorption when lixisenatide as monotherapy is administered subcutaneously in the abdomen, deltoid, or thigh.
The apparent volume of distribution of insulin glargine after subcutaneous administration of the insulin glargine/lixisenatide combinations (Vss/F) is approximately 1700 L.
Lixisenatide has a low level (55%) of binding to human proteins. The apparent volume of distribution of lixisenatide after subcutaneous administration of insulin glargine/lixisenatide combinations (Vz/F) is approximately 100 L.
A metabolism study in diabetic patients who received insulin glargine alone indicates that insulin glargine is rapidly metabolised at the carboxyl terminus of the B chain to form two active metabolites, M1 (21A-Gly-insulin) and M2 (21A-Gly-des-30B-Thr-insulin). In plasma, the principal circulating compound is the metabolite M1. The pharmacokinetic and pharmacodynamic findings indicate that the effect of the subcutaneous injection with insulin glargine is principally based on exposure to M1.
As a peptide, lixisenatide is eliminated through glomerular filtration, followed by tubular reabsorption and subsequent metabolic degradation, resulting in smaller peptides and amino acids, which are reintroduced in the protein metabolism.
After single subcutaneous administration of the insulin glargine/lixisenatide combination, the mean apparent clearance (CL/F) of insulin glargine was approximately 120 L/h.
After multiple-dose subcutaneous administration of Lixisenatide in patients with type 2 diabetes, mean terminal half-life was approximately 3 hours and the mean apparent clearance (CL/F) about 35 L/h.
In subjects with mild (creatinine clearance calculated by the Cockcroft-Gault formula 60-90 ml/min), moderate (creatinine clearance 30-60 ml/min) and severe renal impairment (creatinine clearance 15-30 ml/min) AUC of lixisenatide was increased by 46%, 51% and 87%, respectively.
Insulin glargine has not been studied in patients with renal impairment. In patients with renal impairment, however, insulin requirements may be diminished due to reduced insulin metabolism.
As lixisenatide is cleared primarily by the kidney, no pharmacokinetic study has been performed in patients with acute or chronic hepatic impairment. Hepatic dysfunction is not expected to affect the pharmacokinetics of lixisenatide.
Insulin glargine has not been studied in diabetes patients with hepatic impairment. In patients with hepatic impairment, insulin requirements may be diminished due to reduced capacity for gluconeogenesis and reduced insulin metabolism.
Insulin glargine:
Effect of age, race, and gender on the pharmacokinetics of insulin glargine has not been evaluated. In controlled clinical studies in adults with insulin glargine (100 units/ml), subgroup analyses based on age, race, and gender did not show differences in safety and efficacy.
Lixisenatide:
Age has no clinically relevant effect on the pharmacokinetics of lixisenatide. In a pharmacokinetic study in elderly non-diabetic subjects, administration of lixisenatide 20 mcg resulted in a mean increase of lixisenatide AUC by 29% in the elderly population (11 subjects aged 65 to 74 years and 7 subjects aged ≥75 years) compared to 18 subjects aged 18 to 45 years, likely related to reduced renal function in the older age group.
Ethnic origin had no clinically relevant effect on the pharmacokinetics of lixisenatide based on the results of pharmacokinetic studies in Caucasian, Japanese and Chinese subjects.
Gender has no clinically relevant effect on the pharmacokinetics of lixisenatide
Body weight has no clinically relevant effect on lixisenatide AUC.
In the presence of anti-lixisenatide antibodies, lixisenatide exposure and variability in exposure are markedly increased regardless of the dose level.
No studies have been performed with Suliqua in children and adolescents below 18 years of age.
No animal studies have been conducted with the combination of insulin glargine and lixisenatide to evaluate repeated dose toxicity, carcinogenesis, genotoxicity, or toxicity to reproduction.
Non-clinical data for insulin glargine reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction.
In 2-year subcutaneous carcinogenicity studies, non-lethal C-cell thyroid tumours were seen in rats and mice and are considered to be caused by a non-genotoxic GLP-1 receptor-mediated mechanism to which rodents are particularly sensitive. C-cell hyperplasia and adenoma were seen at all doses in rats and a no observed adverse effect level (NOAEL) could be not defined. In mice, these effects occurred at exposure ratio above 9.3-fold when compared to human exposure at the therapeutic dose. No C-cell carcinoma was observed in mice and C-cell carcinoma occurred in rats with an exposure ratio relative to exposure at human therapeutic dose of about 900-fold.
In 2-year subcutaneous carcinogenicity study in mice, 3 cases of adenocarcinoma in the endometrium were seen in the mid dose group with a statistically significant increase, corresponding to an exposure ratio of 97-fold. No treatment-related effect was demonstrated.
Animal studies did not indicate direct harmful effects with respect to male and female fertility in rats. Reversible testicular and epididymal lesions were seen in dogs treated with lixisenatide. No related effect on spermatogenesis was seen in healthy men.
In embryo-foetal development studies, malformations, growth retardation, ossification retardation and skeletal effects were observed in rats at all doses (5-fold exposure ratio compared to human exposure) and in rabbits at high doses (32-fold exposure ratio compared to human exposure) of lixisenatide. In both species, there was a slight maternal toxicity consisting of low food consumption and reduced body weight. Neonatal growth was reduced in male rats exposed to high doses of lixisenatide during late gestation and lactation, with a slightly increased pup mortality observed.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.