SPRAVATO Nasal spray, solution Ref.[10648] Active ingredients: Esketamine
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Psychoanaleptics; Other antidepressants
ATC code: N06AX27
Mechanism of action
Esketamine is the S-enantiomer of racemic ketamine. It is a non-selective, non-competitive, antagonist of the N-methyl-D-aspartate (NMDA) receptor, an ionotropic glutamate receptor. Through NMDA receptor antagonism, esketamine produces a transient increase in glutamate release leading to increases in α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) stimulation and subsequently to increases in neurotrophic signalling which may contribute to the restoration of synaptic function in these brain regions involved with the regulation of mood and emotional behaviour. Restoration of dopaminergic neurotransmission in brain regions involved in the reward and motivation, and decreased stimulation of brain regions involved in anhedonia, may contribute to the rapid response.
Pharmacodynamic effects
Abuse potential
In a study of abuse potential conducted in recreational polydrug users (n=41), single doses of esketamine nasal spray (84 mg and 112 mg) and the positive control drug intravenous ketamine (0.5 mg/kg infused over 40 minutes) produced significantly greater scores than placebo on subjective ratings of "drug liking" and on other measures of subjective drug effects.
Clinical efficacy and safety
The efficacy and safety of esketamine nasal spray was investigated in five Phase 3 clinical studies (TRD3001, TRD3002, TRD3003, TRD3004, and TRD3005) in adult patients (18 to 86 years) with treatment-resistant depression (TRD) who met DSM-5 criteria for major depressive disorder and were non-responders to at least two oral antidepressants (ADs) treatments, of adequate dosage and duration, in the current major depressive episode. 1 833 adult patients were enrolled, of which 1 601 patients were exposed to esketamine. Additionally, 202 patients were randomised (122 patients received esketamine) in Phase 2 study TRD2005 in Japan, 252 patients were randomised (126 patients received esketamine) in Phase 3 study TRD3006 primarily in China, and 676 patients were randomised (334 patients received esketamine) in Phase 3 study TRD3013.
The efficacy and safety of esketamine nasal spray was investigated in two Phase 3 clinical studies in adult patients (18 to 64 years) with moderate to severe MDD (MADRS total score >28) who had affirmative responses to Mini International Neuropsychiatric Interview (MINI) questions B3 ("Think [even momentarily] about harming or of hurting or of injuring yourself: with at least some intent or awareness that you might die as a result; or think about suicide [i.e., about killing yourself]?") and B10 ("Intend to act on thoughts of killing yourself in the past 24 hours?"). 456 adult patients were enrolled, of which 227 patients were exposed to Spravato.
Treatment-resistant depression – Short-term studies
Esketamine was evaluated in three Phase 3 short-term (4-week) randomised, double-blind, active-controlled studies in patients with TRD. Studies TRANSFORM-1 (TRD3001) and TRANSFORM-2 (TRD3002) were conducted in adults (18 to <65 years) and Study TRANSFORM-3 (TRD3005) was conducted in adults ≥65 years of age. Patients in TRD3001 and TRD3002 initiated treatment with esketamine 56 mg plus a newly initiated daily oral AD or a newly initiated daily oral AD plus placebo nasal spray on day 1. Esketamine dosages were then maintained on 56 mg or titrated to 84 mg or matching placebo nasal spray administered twice-weekly during a 4-week double-blind induction phase. Esketamine doses of 56 mg or 84 mg were fixed in Study TRD3001 and flexible in Study TRD3002. In Study TRD3005, patients (≥65 years) initiated treatment with Esketamine 28 mg plus a newly initiated daily oral AD or a newly initiated daily oral AD plus placebo nasal spray (day 1). Esketamine dosages were titrated to 56 mg or 84 mg or matching placebo nasal spray administered twice-weekly during a 4-week double-blind induction phase. In the flexible dose studies, TRD3002 and TRD3005, up titration of esketamine dose was based on clinical judgement and dose could be down titrated based on tolerability. A newly initiated open-label oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) was initiated on day 1 in all studies. The selection of the newly initiated oral AD was determined by the investigator based on the patient's prior treatment history. In all short-term studies, the primary efficacy endpoint was change in MADRS total
score from baseline to day 28.
Baseline demographic and disease characteristics for patient in TRD3002, TRD3001, and TRD3005 are presented in Table 4.
Table 4. Baseline demographic characteristics for TRD3002, TRD3001, and TRD3005 (full analysis sets):
| Study TRD3002 (N=223) | Study TRD3001 (N=342) | Study TRD3005 (N=137) | |
|---|---|---|---|
| Age, years | |||
| Median (Range) | 47.0 (19; 64) | 47.0 (18; 64) | 69.0 (65; 86) |
| Sex, n (%) | |||
| Male | 85 (38.1%) | 101 (29.5%) | 52 (38.0%) |
| Female | 138 (61.9%) | 241 (70.5%) | 85 (62.0%) |
| Race, n (%) | |||
| White | 208 (93.3%) | 262 (76.6%) | 130 (94.9%) |
| Black or African American | 11 (4.9%) | 19 (5.6%) | -- |
| Prior oral antidepressants with nonresponse (i.e., failed antidepressants) | |||
| Number of specific antidepressants, n (%) | |||
| 2 | 136 (61.0%) | 167 (48.8%) | 68 (49.6%) |
| 3 or more | 82 (36.8%) | 167 (48.8%) | 58 (42.3%) |
| Newly initiated oral antidepressant medication initiated at randomisation, n (%) | |||
| SNRI | 152 (68.2%) | 196 (57.3%) | 61 (44.5%) |
| SSRI | 71 (31.8%) | 146 (42.7%) | 76 (55.5%) |
| Withdrawn from study (for any reason), n/N (%) | 30/227 (13.2%) | 31/346 (9.0%) | 16/138 (11.6%) |
In the flexible dose study TRD3002, at day 28, 67% of the patients randomised to Spravato were on 84 mg. In study TRD3002, esketamine plus a newly initiated oral AD demonstrated clinically meaningful and statistical superiority compared to a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray (Table 5), and symptom reduction was observed as early as 24 hours post-dose.
In study TRD3001, a clinically meaningful treatment effect in change in MADRS total scores from baseline at the end of the 4-week induction phase was observed favouring esketamine plus newly initiated oral AD compared with a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray (Table 5). In Study TRD3001, the treatment effect for the esketamine 84 mg plus oral AD group compared with oral AD plus placebo was not statistically significant.
In study TRD3005, at day 28, 64% of the patients randomised to esketamine were on 84 mg, 25% on 56 mg, and 10% on 28 mg. In study TRD3005, a clinically meaningful but not statistically significant treatment effect in change in MADRS total scores from baseline at the end of the 4-week induction phase was observed favouring esketamine plus newly initiated oral AD compared with a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray (Table 5). Subgroup analyses suggest limited efficacy in the population over 75 years old.
Table 5. Primary efficacy results for change in MADRS total score for 4-week clinical trials (ANCOVA BOCF*):
| Study no. | Treatment group§ | Number of patients | Mean baseline score (SD) | LS mean change from baseline to end of week 4 (SE) | LS mean difference (95% CI)† |
|---|---|---|---|---|---|
| TRD3001 | Spravato 56 mg + oral AD | 115 | 37.4 (4.8) | -18.9 (1.3) | -4.3 (-7.8, -0.8)# |
| Spravato 84 mg + oral AD | 114 | 37.8 (5.6) | -16.2 (1.3) | -1.2 (-4.7, 2.3)# | |
| Oral AD + placebo nasal spray | 113 | 37.5 (6.2) | -14.7 (1.3) | ||
| TRD3002 | Spravato (56 mg or 84 mg) + oral AD | 114 | 37.0 (5.7) | -17.7 (1.3) | -3.5 (-6.7, -0.3)‡ |
| Oral AD + placebo nasal spray | 109 | 37.3 (5.7) | -14.3 (1.3) | ||
| TRD3005 (≥65 years) | Spravato (28 mg, 56 mg or 84 mg) + oral AD | 72 | 35.5 (5.9) | -10.1 (1.7) | -2.9 (-6.5, 0.6)# |
| Oral AD + placebo nasal spray | 65 | 34.8 (6.4) | -6.8 (1.7) |
SD = standard deviation; SE = standard error; LS Mean = least-squares mean; CI = confidence interval; AD = antidepressant
* ANCOVA analysis using Baseline Observation Carried Forward, which means that for a patient who discontinues from treatment, it is assumed that the depression level returns to the baseline level (i.e. the depression level is the same as before start of treatment)
§ Nasally administered esketamine or placebo; oral AD = a newly initiated AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline)
† Difference (Spravato + oral AD minus Oral AD + placebo nasal spray) in least-squares mean change from baseline
‡ Treatment group that was statistically significantly superior to Oral AD + placebo nasal spray
# Median unbiased estimate (i.e., weighted combination of the LS means of the difference from Oral AD + placebo nasal spray), and 95% flexible confidence interval
Response and remission rates
Response was defined as ≥50% reduction in the MADRS total score from baseline of the induction phase. Based on the reduction in MADRS total score from baseline, the proportion of patients in Studies TRD3001, TRD3002 and TRD3005 who demonstrated response to esketamine plus oral AD treatment was greater than for oral AD plus placebo nasal spray throughout the 4-week double-blind induction phase (Table 6).
Remission was defined as a MADRS total score ≤12. In all three studies, a greater proportion of patients treated with esketamine plus oral AD were in remission at the end of the 4-week double-blind induction phase than for oral AD plus placebo nasal spray (Table 6).
Table 6. Response and remission rates in 4-week clinical trials based on BOCF* data:
| Study No. | Treatment group§ | Number of patients (%) | |||||
|---|---|---|---|---|---|---|---|
| Response rate† | Remission rate‡ | ||||||
| 24 hours | Week 1 | Week 2 | Week 3 | Week 4 | Week 4 | ||
| TRD3001 | Spravato 56 mg + oral AD | 20 (17.4%) | 21 (18.3%) | 29 (25.2%) | 52 (45.2%) | 61 (53.0%) | 40 (34.8%) |
| Spravato 84 mg + oral AD | 17 (14.9%)# | 16 (14.0%) | 25 (21.9%) | 33 (28.9%) | 52 (45.6%) | 38 (33.3%) | |
| Oral AD + placebo nasal spray | 8 (7.1%) | 5 (4.4%) | 15 (13.3%) | 25 (22.1%) | 42 (37.2%) | 33 (29.2%) | |
| TRD3002 | Spravato 56 mg or 84 mg + oral AD | 18 (15.8%) | 15 (13.2%) | 29 (25.4%) | 54 (47.4%) | 70 (61.4%) | 53 (46.5%) |
| Oral AD + placebo nasal spray | 11 (10.1%) | 13 (11.9%) | 23 (21.1%) | 35 (32.1%) | 52 (47.7%) | 31 (28.4%) | |
| TRD3005 (≥65 years) | Spravato 28 mg, 56 mg or 84 mg + oral AD | NA | 4 (5.6%) | 4 (5.6%) | 9 (12.5%) | 17 (23.6%) | 11 (15.3%) |
| Oral AD + placebo nasal spray | NA | 3 (4.6%) | 8 (12.3%) | 8 (12.3%) | 8 (12.3%) | 4 (6.2%) | |
AD = antidepressant; NA = not available
* Baseline Observation Carried Forward, which means that for a patient who discontinues from treatment, it is assumed that the depression level returns to the baseline level (i.e. the depression level is the same as before start of treatment).
§ Nasally administered Spravato or placebo; oral AD = a newly initiated AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline)
† Response was defined as ≥50% reduction in the MADRS total score from baseline
‡ Remission was defined as MADRS total score ≤12
# First dose was Spravato 56 mg + oral AD
Treatment-resistant depression – Long-term studies
Relapse-prevention study
The maintenance of antidepressant efficacy was demonstrated in a relapse prevention trial. Study SUSTAIN-1 (TRD3003) was a long-term randomised, double-blind, parallel-group, active-controlled, multicentre, relapse prevention study. The primary outcome measure to assess the prevention of depressive relapse was measured as time to relapse. Overall a total of 705 patients were enrolled; 437 directly enrolled; 150 transferred from TRD3001, and 118 transferred from TRD3002. Patients directly enrolled were administered esketamine (56 mg or 84 mg twice weekly) plus oral AD in a 4-week open label induction phase. At the end of the open label induction phase, 52% of patients were in remission (MADRS total score ≤12) and 66% of patients were responders (≥50% improvement in MADRS total score). Patients who were responders (455), continued receiving treatment with esketamine plus oral AD in a 12-week optimisation phase. After the induction phase, patients received esketamine weekly for 4 weeks and starting from week 8, an algorithm (based on the MADRS) was used to determine the dosing frequency; patients in remission (i.e., MADRS total score was ≤12) were dosed every other week, however, if the MADRS total score increased to >12, then the frequency was increased to weekly dosing for the next 4 weeks; with the objective of maintaining the patient on the lowest dosing frequency to maintain response/remission. At the end of 16 weeks of treatment period, patients in stable remission (n=176) or stable response (n=121) were randomised to continue with esketamineor stop esketamine and switch to placebo nasal spray. Stable remission was defined as MADRS total score ≤12 in at least 3 of the last 4 weeks of the optimisation phase and stable response was defined as ≥50% reduction in the MADRS total score from baseline for the last 2 weeks of the optimisation phase, but not in stable remission.
Stable remission
Patients in stable remission who continued treatment with Spravato plus oral AD experienced a statistically significantly longer time to relapse of depressive symptoms than did patients on a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray (Figure 1). Relapse was defined as a MADRS total score ≥22 for 2 consecutive weeks or hospitalisation for worsening depression or any other clinically relevant event indicative of relapse. The median time to relapse for a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray group was 273 days, whereas the median was not estimable for Spravato plus oral AD, as this group never reached 50% relapse rate.
Figure 1. Time to relapse in patients in stable remission in study TRD3003 (full analysis set):
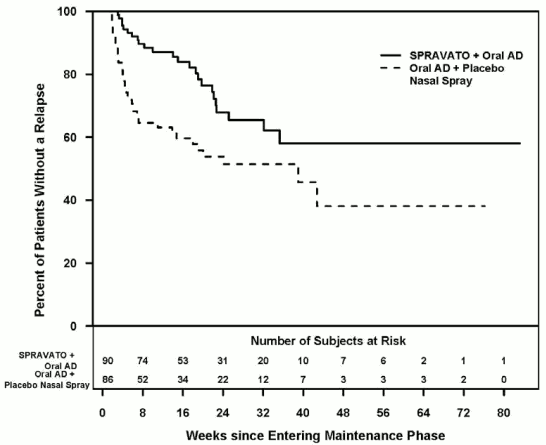
For patients in stable remission, the relapse rate based on Kaplan-Meier estimates during the 12- and 24-weeks double-blind follow up period was 13% and 32% for esketamine and 37% and 46% for placebo nasal spray, respectively.
Stable response
he efficacy results were also consistent for patients in stable response who continued treatment with esketamine plus oral AD; patients experienced a statistically significantly longer time to relapse of depressive symptoms than did patients on a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray (Figure 2). The median time to relapse for a newly initiated oral AD (SNRI: duloxetine, venlafaxine extended release; SSRI: escitalopram, sertraline) plus placebo nasal spray group (8 days) was shorter compared to esketamine plus oral AD group (635 days).
Figure 2. Time to relapse in patients in stable response in study TRD3003 (full analysis set):
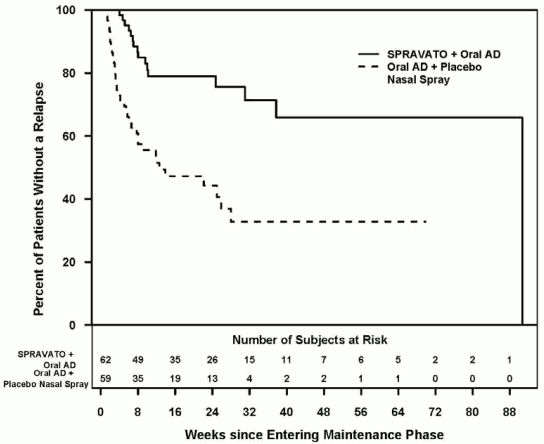
For patients in stable response, the relapse rate based on Kaplan-Meier estimates during the 12- and 24-weeks double-blind follow up period was 21% and 21% for esketamine and 47% and 56% for placebo nasal spray, respectively.
Enrollment in TRD3003 was staggered over approximately 2 years. The maintenance phase was of variable duration and continued until the individual patient had a relapse of depressive symptoms or discontinued for any other reason, or the study ended because the required number of relapse events occurred. Exposure numbers were influenced by the study stopping at a pre-determined number of relapses based on the interim analysis. After an initial 16 weeks of treatment with esketamine plus oral AD, the median duration of exposure to esketamine in the maintenance phase was 4.2 months (range: 1 day to 21.2 months) in Spravato-treated patients (stable remission and stable response). In this study, 31.6% of patients received esketamine for greater than 6 months and 7.9% of patients received esketamine for greater than 1 year in the maintenance phase.
Dosing frequency
The dosing frequency used the majority of the time during the maintenance phase is shown in Table 7. Of the patients randomised to Spravato, 60% received 84 mg and 40% received 56 mg dose.
Table 7. Dosing frequency used the majority of the time; maintenance phase (Study TRD3003):
| Stable Remission | Stable Responders | |||
|---|---|---|---|---|
| Spravato + Oral AD (N=90) | Oral AD + Placebo Nasal Spray (N=86) | Spravato + Oral AD (N=62) | Oral AD + Placebo Nasal Spray (N=59) | |
| Majority dosing frequency | ||||
| Weekly | 21 (23.3%) | 27 (31.4%) | 34 (54.8%) | 36 (61.0%) |
| Every other week | 62 (68.9%) | 48 (55.8%) | 21 (33.9%) | 19 (32.2%) |
| Weekly or every other week | 7 (7.8%) | 11 (12.8%) | 7 (11.3%) | 4 (6.8%) |
Study TRD3013 (ESCAPE-TRD)
The efficacy of Spravato was evaluated in a long-term randomised, open-label, rater-blinded, active-controlled study (TRD3013) where esketamine was compared with quetiapine prolonged/extended-release (XR) in 676 adult patients (18-74 years) with TRD who continued to take their current oral AD (an SSRI or SNRI). Patients received treatment with flexibly dosed esketamine (28, 56, or 84 mg) or quetiapine XR, in line with the dosing recommendations in the SmPCs in use at the time of study initiation.
The primary efficacy endpoint was remission (MADRS total score of ≤10) at Week 8 and the key secondary endpoint was remaining relapse-free through Week 32 after remission at Week 8. Relapse was defined as a MADRS total score ≥22 for 2 consecutive weeks or hospitalisation for worsening depression or any other clinically relevant event indicative of relapse.
The baseline demographic and disease characteristics of patients were similar between the esketamine plus oral AD and quetiapine XR plus oral AD groups. The mean (SD) baseline MADRS total scores were 31.4 (6.06) for the esketamine plus oral AD group and 31.0 (5.83) for the quetiapine XR plus oral AD group.
Esketamine plus oral AD demonstrated clinically meaningful and statistical superiority compared to quetiapine XR plus oral AD on both the primary (Table 8) and key secondary (Table 9) efficacy measure.
Table 8. Primary efficacy results for TRD3013 Studya:
| Treatment group | Spravato + oral AD | Quetiapine XR + oral AD |
|---|---|---|
| Number of patients in remission at Week 8 | 91/336 (27.1%) | 60/340 (17.6%) |
| Adjusted risk difference in percentage (95% CI)b | 9.5 (3.3, 15.8) | – |
| P-valuec | P=0.003 | – |
CI = confidence interval; AD = antidepressant; XR = extended release
a A patient who discontinued study intervention before Week 8 was considered as a negative outcome (i.e. non-remission). For patients for whom no MADRS result was available at the Week 8 visit but who did not discontinue study intervention or withdraw from study before Week 8, LOCF of MADRS was applied.
b Mantel-Haenszel estimate of the risk difference, stratified by age groups (18-64; ≥65) and total number of treatment failures is used. This estimated difference indicates an advantage for esketamine.
c Cochran–Mantel–Haenszel (CMH) test, adjusting for age groups (18-64; ≥65) and total number of treatment failures.
Table 9. Key secondary efficacy results for TRD3013 Studya:
| Treatment group | Spravato + oral AD | Quetiapine XR + oral AD |
|---|---|---|
| Number of patients both in remission at Week 8 and relapse-free at Week 32 | 73/336 (21.7%) | 48/340 (14.1%) |
| Adjusted risk difference in percentage (95% CI)b | 7.7 (2.0, 13.5) | – |
| P-valuec | P=0.008 | – |
CI = confidence interval; AD = antidepressant; XR = extended release
a A patient who discontinued study intervention was considered as a negative outcome. For patients for whom no MADRS result was available at the Week 8 visit but who did not discontinue study intervention or withdraw from study before Week 8, LOCF of MADRS was applied.
b Mantel-Haenszel estimate of the risk difference, stratified by age groups (18-64; ≥65) and total number of treatment failures is used. This estimated difference indicates an advantage for esketamine.
c Cochran–Mantel–Haenszel (CMH) test, adjusting for age groups (18-64; ≥65) and total number of treatment failures.
Treatment discontinuation rates over the 32-week treatment period due to adverse events, lack of efficacy, and overall were 4.2%, 8.3%, and 23.2% respectively for patients in the esketamine plus oral AD group and 11.5%, 15.0%, and 40.3% respectively for patients in the quetiapine XR plus oral AD group.
Treatment-resistant depression – Short-term study in Japanese patients
The efficacy of Spravato was also evaluated in a short-term (4-week) randomised, double-blind, active-controlled study (TRD2005) in 202 adult Japanese patients with TRD. Patients received 4 weeks of induction treatment with esketamine fixed-dose of 28 mg, 56 mg, 84 mg or placebo nasal spray in addition to continued current oral AD. The primary efficacy endpoint was change in MADRS total score from baseline to day 28. The baseline demographic and disease characteristics of patients were similar between the esketamine plus AD and placebo nasal spray plus AD groups.
In study TRD2005, no statistically significant difference in change in MADRS total scores from baseline at the end of the 4-week induction phase was observed for any of the esketamine plus oral AD dosages compared with oral AD plus placebo nasal spray (Table 10).
Table 10. Primary efficacy results for change in MADRS total score for 4-week TRD2005 Study in Japanese patients (MMRM):
| Treatment group | Number of patients | Mean baseline score (SD) | LS mean change from baseline to end of week 4 (SE) | LS mean difference (90% CI)†,# |
|---|---|---|---|---|
| Spravato 28 mg + oral AD | 41 | 38.4 (6.1) | -15.6 (1.8) | -1.0 -5.77; 3.70 |
| Spravato 56 mg + oral AD | 40 | 37.9 (5.4) | -14.0 (1.9) | 0.6 -4.32; 5.47 |
| Spravato 84 mg + oral AD | 41 | 35.9 (5.3) | -15.5 (1.8) | -0.9 -5.66; 3.83 |
| Oral AD + placebo nasal spray | 80 | 37.7 (5.7) | -14.6 (1.3) |
SD = standard deviation; SE = standard error; LS Mean = least-squares mean; CI = confidence interval; AD = antidepressant.
† Difference (Spravato + oral AD minus Oral AD + placebo nasal spray) in least-squares mean change from baseline.
# Confidence interval is based on the Dunnett adjustment.
Treatment-resistant depression – Short-term study in Chinese patients
The efficacy of Spravato was also evaluated in a short-term (4-week) randomised, double-blind, active-controlled study (TRD3006) in 252 adult patients (224 Chinese patients, 28 non-Chinese patients) with TRD.
Patients received 4 weeks of induction treatment with flexibly dosed esketamine (56 mg or 84 mg) or placebo nasal spray, in addition to a newly initiated oral AD. The primary efficacy endpoint was change in MADRS total score from baseline to day 28. The baseline demographic and disease characteristics of patients were similar between the esketamine plus AD and placebo nasal spray plus AD groups.
In study TRD3006, no statistically significant difference in change in MADRS total scores from baseline at the end of the 4-week induction phase was observed for esketamine plus oral AD compared with oral AD plus placebo nasal spray (Table 11).
Table 11. Primary efficacy results for change in MADRS total score for 4-week TRD3006 Study (MMRM):
| Treatment group | Number of patients# | Mean baseline score (SD) | LS mean change from baseline to end of week 4 (SE) | LS mean difference (95% CI)† |
|---|---|---|---|---|
| All patients | ||||
| Spravato (56 mg or 84 mg) + oral AD | 124 | 36.5 (5.21) | -11.7 (1.09) | -2.0 -4.64; 0.55 |
| Oral AD + placebo nasal spray | 126 | 35.9 (4.50) | -9.7 (1.09) | |
| Chinese population | ||||
| Spravato (56 mg or 84 mg) + oral AD | 110 | 36.2 (5.02) | -8.8 (0.95) | -0.7 -3.35; 1.94 |
| Oral AD + placebo nasal spray | 112 | 35.9 (4.49) | -8.1 (0.95) | |
SD = standard deviation; SE = standard error; LS Mean = least-squares mean; CI = confidence interval; AD = antidepressant.
# Two patients did not receive oral AD and were not included in the efficacy analysis.
† Difference (Spravato + oral AD minus Oral AD + placebo nasal spray) in least-squares mean change from baseline.
Acute short-term treatment of psychiatric emergency due to Major Depressive Disorder
Spravato was investigated in two identical Phase 3 short-term (4-week) randomised, double-blind, multicentre, placebo-controlled studies, Aspire I (SUI3001) and Aspire II (SUI3002) in adult patients with moderate to severe MDD (MADRS total score >28) who had affirmative responses to MINI questions B3 ("Think [even momentarily] about harming or of hurting or of injuring yourself: with at least some intent or awareness that you might die as a result; or think about suicide [i.e., about killing yourself]?") and B10 ("Intend to act on thoughts of killing yourself in the past 24 hours?"). In these studies, patients received treatment with esketamine 84 mg or placebo nasal spray twice-weekly for 4 weeks. All patients received comprehensive standard of care (SOC) treatment, including an initial inpatient hospitalisation and a newly initiated or optimised oral antidepressant (AD) therapy (AD monotherapy or AD plus augmentation) as determined by the investigator. In the physician's opinion, acute psychiatric hospitalisation was clinically warranted due to the subject's immediate risk of suicide. After the first dose, a one-time dose reduction to esketamine 56 mg was allowed for patients unable to tolerate the 84 mg dose.
The baseline demographic and disease characteristics of patients in SUI3001 and SUI3002 were similar between the esketamine plus SOC or placebo nasal spray plus SOC groups. The median patient age was 40 years (range 18 to 64 years), 61% were female; 73% Caucasian and 6% Black; and 63% of patients had at least one prior suicide attempt. Prior to entering the study, 92% of the patients were receiving antidepressant therapy. During the study, as part of standard of care treatment, 40% of patients received AD monotherapy, 54% of patients received AD plus augmentation regimen, and 6% received both AD monotherapy/AD plus augmentation regimen.
The primary efficacy measure was the reduction of symptoms of MDD as measured by the change from baseline MADRS total score at 24 hours after first dose (Day 2).
In SUI3001 and SUI3002, Spravato plus SOC demonstrated statistical superiority on the primary efficacy measure compared to placebo nasal spray plus SOC (see Table 12).
Table 12. Primary efficacy results for change from baseline in MADRS total score at 24 hours after first dose (Studies SUI3001 and SUI3002) (ANCOVA BOCF*):
| Study No. | Treatment Group‡ | Number of Patients | Mean Baseline Score (SD) | LS Mean Change from Baseline to 24 hr Post First Dose (SE) | LS Mean Difference (95% CI)§ |
|---|---|---|---|---|---|
| Study 1 (SUI3001) | Spravato 84 mg + SOC | 112 | 41.2 (5.87) | -15.7 (1.05) | -3.7 (-6.41; -0.92)# P=0.006 |
| Placebo nasal spray + SOC | 112 | 41.0 (6.29) | -12.1 (1.03) | – | |
| Study 2 (SUI3002) | Spravato 84 mg + SOC | 114 | 39.5 (5.19) | -15.9 (1.02) | -3.9 (-6.65; -1.12)# P=0.006 |
| Placebo nasal spray + SOC | 113 | 39.9 (5.76) | -12.0 (1.06) | – | |
| Pooled Studies 1 and 2 | Spravato 84 mg + SOC | 226 | 40.3 (5.60) | -15.8 (0.73) | -3.8 (-5.69; -1.82) |
| Placebo nasal spray + SOC | 225 | 40.4 (6.04) | -12.1 (0.73) | – |
SD = standard deviation; SE = standard error; LS Mean = least-squares mean; CI = confidence interval; SOC = standard of care
* ANCOVA analysis using Baseline Observation Carried Forward: In SUI3001, 2 subjects (1 subject in each group) did not have the Day 2 (24 hours post first dose) MADRS total score and in SUI3002, 6 subjects (4 subjects in Esketamine and 2 subjects in Placebo) did not have the Day 2 (24 hours post first dose) MADRS total score. For these subjects, it is assumed that the depression level returns to the baseline level (i.e., the depression level is the same as the start of treatment) and the MADRS total scores from baseline were carried forward for the analysis
‡ Nasally administered esketamine or placebo
§ Difference (Spravato + SOC minus placebo nasal spray + SOC) in least-squares mean change from baseline
# Treatment groups that were statistically significantly superior to placebo nasal spray + SOC.
The treatment differences (95% CI) in change from baseline in MADRS total score at Day 2 (24 hours post first dose) between esketamine + SOC and placebo + SOC were -4.70 (-7.16; -2.24) for the subpopulation that reported a prior suicide attempt (N=284) and -2.34 (-5.59; 0.91) for the subpopulation that did not report a prior suicide attempt (N=166).
Time course of treatment response
In both SUI3001 and SUI3002, esketamine's treatment difference compared to placebo was observed starting at 4 hours. Between 4 hours and Day 25, the end of the treatment phase, both the esketamine and placebo groups continued to improve; the difference between the groups generally remained but did not appear to increase over time through Day 25. Figure 3 depicts time course of the primary efficacy measure of change in MADRS total score using pooled studies SUI3001 and SUI3002.
Figure 3. Least squares mean change from baseline in MADRS total score over time in SUI3001 and SUI3002* (pooled data, safety analysis set) – ANCOVA BOCF:
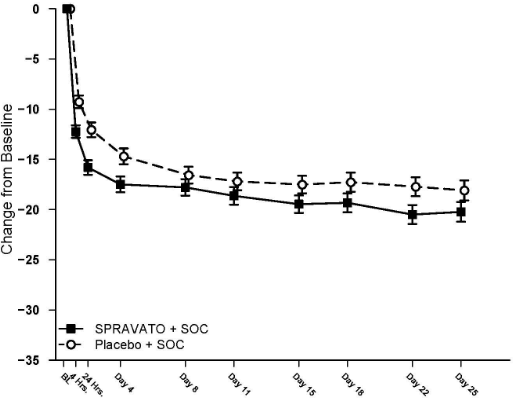
* Note: In these studies, after the first dose, a one-time dose reduction to Spravato 56 mg was allowed for patients unable to tolerate the 84 mg dose. Approximately 16% of patients had reduction in Spravato dosage from 84 mg to 56 mg twice weekly.
Remission rates
In the Phase 3 studies, the percentage of patients who achieved remission (MADRS total score ≤12 at any given time during the study) was greater in the esketamine + SOC group than in the placebo + SOC group at all timepoints during the 4-week double-blind treatment phase (Table 13).
Table 13. Patients who achieved remission of MDD; double-blind treatment phase; full efficacy analysis set:
| SUI3001 | SUI3002 | Pooled Studies (SUI3001 and SUI3002) | ||||
|---|---|---|---|---|---|---|
| Placebo + SOC 112 | Spravato + SOC 112 | Placebo + SOC 113 | Spravato + SOC 114 | Placebo + SOC 225 | Spravato + SOC 226 | |
| Day 1, 4 hours post first dose | ||||||
| Patients with Remission of MDD | 9 (8.0%) | 12 (10.7%) | 4 (3.5%) | 12 (10.5%) | 13 (5.8%) | 24 (10.6%) |
| Day 2, 24 hours post first dose | ||||||
| Patients with Remission of MDD | 10 (8.9%) | 21 (18.8%) | 12 (10.6%) | 25 (21.9%) | 22 (9.8%) | 46 (20.4%) |
| Day 25 (predose) | ||||||
| Patients with Remission of MDD | 38 (33.9%) | 46 (41.1%) | 31 (27.4%) | 49 (43.0%) | 69 (30.7%) | 95 (42.0%) |
| Day 25 (4 hours postdose) | ||||||
| Patients with Remission of MDD | 42 (37.5%) | 60 (53.6%) | 42 (37.2%) | 54 (47.4%) | 84 (37.3%) | 114 (50.4%) |
SOC = standard of care
Note: Remission is based on a MADRS total score of ≤12. Subjects who did not meet such criterion or discontinued prior to the time point for any reason are not considered to be in remission.
Effects on suicidality
Overall patients in both treatment groups experienced improvement in the severity of their suicidality as measured by the Clinical Global Impression – Severity of Suicidality-revised (CGI-SS-r) scale at the 24-hour endpoint, though there was no statistically significant difference between treatment groups.
The long-term efficacy of esketamine to prevent suicide has not been established.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Spravato in the treatment of major depressive disorder in one or more subsets of the paediatric population (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
The mean absolute bioavailability of 84 mg esketamine administered as a nasal spray is approximately 48%.
Esketamine is rapidly absorbed by the nasal mucosa following nasal administration and can be measured in plasma within 7 minutes following a 28 mg dose. The time to reach maximum plasma concentration (tmax) is typically 20 to 40 minutes after the last nasal spray of a treatment session (see section 4.2).
Dose-dependent increases in the maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC∞) of esketamine nasal spray were produced by doses of 28 mg, 56 mg and 84 mg.
The pharmacokinetic profile of esketamine is similar after a single dose and repeat dose administration with no accumulation in plasma when esketamine is administered twice a week.
Distribution
The mean steady-state volume of distribution of esketamine administered by the intravenous route is 709 L.
The proportion of the total concentration of esketamine that is bound to proteins in human plasma is on average 43 to 45%. The degree to which esketamine is bound to plasma proteins is not dependent on hepatic or renal function.
Esketamine is not a substrate of transporters P-glycoprotein (P-gp; multidrug resistance protein 1), breast cancer resistance protein (BCRP), or organic anion transporter (OATP) 1B1, or OATP1B3. Esketamine does not inhibit these transporters or multi-drug and toxin extrusion 1 (MATE1) and MATE2-K, or organic cation transporter 2 (OCT2), OAT1, or OAT3.
Biotransformation
Esketamine is extensively metabolised in the liver. The primary metabolic pathway of esketamine in human liver microsomes is N-demethylation to form noresketamine. The main cytochrome P450 (CYP) enzymes responsible for esketamine N-demethylation are CYP2B6 and CYP3A4. Other CYP enzymes, including CYP2C19 and CYP2C9, contribute to a much smaller extent. Noresketamine is subsequently metabolised via CYP-dependent pathways to other metabolites, some of which undergo glucuronidation.
Elimination
The mean clearance of esketamine administered by the intravenous route was approximately 89 L/hour. After Cmax was reached following nasal administration, the decline in esketamine concentrations in plasma was rapid for the first few hours and then more gradual. The mean terminal half-life following administration as a nasal spray generally ranged from 7 to 12 hours.
Following intravenous administration of radiolabelled esketamine, approximately 78% and 2% of administered radioactivity was recovered in urine and faeces, respectively. Following oral administration of radiolabelled esketamine, approximately 86% and 2% of administered radioactivity was recovered in urine and faeces, respectively. The recovered radioactivity consisted primarily of esketamine metabolites. For the intravenous and oral routes of administration, <1% of the dose was excreted in the urine as unchanged drug.
Linearity/non-linearity
Esketamine exposure increases with dose from 28 mg to 84 mg. The increase in Cmax and AUC values was less than dose-proportional between 28 mg and 56 mg or 84 mg, but it was nearly dose proportional between 56 mg and 84 mg.
Interactions
Effect of other medicinal products on esketamine
Hepatic enzyme inhibitors:
Pre-treatment of healthy subjects with oral ticlopidine, an inhibitor of hepatic CYP2B6 activity, (250 mg twice daily for 9 days prior to and on the day of esketamine administration) had no effect on the Cmax of esketamine administered as a nasal spray. The AUC∞ of esketamine was increased by approximately 29%. The terminal half-life of esketamine was not affected by ticlopidine pre-treatment.
Pre-treatment with oral clarithromycin, an inhibitor of hepatic CYP3A4 activity, (500 mg twice daily for 3 days prior to and on the day of esketamine administration) increase the mean Cmax and AUC∞ of nasally administered esketamine by approximately 11% and 4%, respectively. The terminal half-life of esketamine was not affected by clarithromycin pre-treatment.
Hepatic enzyme inducers:
Pre-treatment with oral rifampicin, a potent inducer of the activity of multiple hepatic CYP enzymes such as CYP3A4 and CYP2B6, (600 mg daily for 5 days prior to esketamine administration) decreased the mean Cmax and AUC∞ values of esketamine administered as a nasal spray by approximately 17% and 28%, respectively.
Other nasal spray products:
Pre-treatment of subjects with a history of allergic rhinitis and pre-exposed to grass pollen with oxymetazoline administered as a nasal spray (2 sprays of 0.05% solution administered at 1 hour prior to nasal administration of esketamine) had minor effects on the pharmacokinetics of esketamine.
Pre-treatment of healthy subjects with nasal administration of mometasone furoate (200 mcg per day for 2 weeks with the last mometasone furoate dose administered at 1 hour prior to nasal administration of esketamine) had minor effects on the pharmacokinetics of esketamine.
Effect of esketamine on other medicinal products
Nasal administration of 84 mg esketamine twice a week for 2 weeks reduced the mean plasma AUC∞ of oral midazolam (single 6 mg dose), a substrate of hepatic CYP3A4, by approximately 16%.
Nasal administration of 84 mg esketamine twice a week for 2 weeks did not affect the mean plasma AUC of oral bupropion (single 150 mg dose), a substrate of hepatic CYP2B6.
Special populations
Elderly (65 years of age and older)
The pharmacokinetics of esketamine administered as a nasal spray was compared between elderly but otherwise healthy subjects and younger healthy adults. The mean esketamine Cmax and AUC∞ values produced by a 28-mg dose were 21% and 18% higher, respectively, in elderly subjects (age range 65 to 81 years) compared with younger adult subjects (age range 22 to 50 years). The mean esketamine Cmax and AUC∞ values produced by an 84-mg dose were 67% and 38% higher in elderly subjects (age range 75 to 85 years) compared with younger adult subjects (age range 24 to 54 years). The terminal half-life of esketamine was similar in the elderly and younger adult subjects (see section 4.2).
Renal impairment
Relative to the subjects with normal renal function (creatinine clearance [CLCR], 88 to 140 mL/min), the Cmax of esketamine was on average 20 to 26% higher in subjects with mild (CLCR, 58 to 77 mL/min), moderate (CLCR, 30 to 47 mL/min), or severe (CLCR, 5 to 28 mL/min, not on dialysis) renal impairment following administration of a 28-mg dose of esketamine nasal spray. The AUC∞ was 13 to 36% higher in the subjects with mild to severe renal impairment.
There is no clinical experience with esketamine administered as a nasal spray in patients on dialysis.
Hepatic impairment
The Cmax and AUC∞ of esketamine produced by a 28-mg doses were similar between subjects with Child-Pugh class A (mild) hepatic impairment and healthy subjects. The Cmax and AUC∞ of esketamine were 8% higher and 103% higher, respectively, in subjects with Child-Pugh class B (moderate) hepatic impairment, relative to healthy subjects.
There is no clinical experience with esketamine administered as a nasal spray in patients with Child-Pugh class C (severe) hepatic impairment (see section 4.2 and 4.4).
Race
The pharmacokinetics of esketamine nasal spray was compared between healthy Asian subjects and Caucasian subjects. Mean plasma esketamine Cmax and AUC∞ values produced by a single, 56 mg dose of esketamine were approximately 14% and 33% higher, respectively, in Chinese subjects compared to Caucasians. On average, esketamine Cmax was 10% lower and AUC∞ was 17% higher in Korean subjects, relative to Caucasian subjects. A population pharmacokinetic analysis was conducted that included Japanese patients with treatment-resistant depression, in addition to healthy Japanese subjects. Based on this analysis, for a given dose, the plasma esketamine Cmax and AUC24h in Japanese subjects were approximately 20% higher relative to non-Asian subjects. The mean terminal half-life of esketamine in the plasma of Asian subjects ranged from 7.1 to 8.9 hours and was 6.8 hours in Caucasian subjects.
Gender and body weight
No significant differences in the pharmacokinetics of esketamine nasal spray were observed for gender and total body weight (>39 to 170 kg) based on population PK analysis.
Allergic rhinitis
The pharmacokinetics of a single, 56 mg dose of esketamine administered as a nasal spray was similar in subjects with allergic rhinitis who were exposed to grass pollen compared to healthy subjects.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of repeated dose toxicity, genotoxicity, neurotoxicity, reproductive toxicity, and carcinogenic potential. Animal studies with ketamine showed evidence of developmental neurotoxicity. The potential for esketamine to have neurotoxic effects on developing foetuses cannot be excluded (see section 4.6).
Genotoxicity
Esketamine was not mutagenic with or without metabolic activation in the Ames test. Genotoxic effects with esketamine were seen in a screening in vitro micronucleus test in the presence of metabolic activation. However, intravenously-administered esketamine was devoid of genotoxic properties in an in vivo bone marrow micronucleus test in rats and an in vivo Comet assay in rat liver cells.
Reproductive toxicity
In an embryo foetal developmental toxicity study with nasally administered ketamine in rats, the offspring was not adversely affected in the presence of maternal toxicity at doses resulting in exposure up to 6-fold higher than human exposure, based on AUC values. In an embryo foetal developmental toxicity study with nasally administered ketamine in rabbits, skeletal malformations were observed and foetal body weight was reduced at maternally toxic doses. Exposure in rabbits was in the region of human exposure based on AUC values.
Published studies in animals (including primates) at doses resulting in light to moderate anaesthesia demonstrate that the use of anaesthetic agents during the period of rapid brain growth or synaptogenesis results in cell loss in the developing brain, that can be associated with prolonged cognitive deficiencies. The clinical significance of these non-clinical findings in not known.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.