TAVNEOS Hard capsule Ref.[28343] Active ingredients: Avacopan
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: Vifor Fresenius Medical Care Renal Pharma France, 100–101 Terrasse Boieldieu, Tour Franklin La Défense 8, 92042 Paris la Défense Cedex, France
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: not yet assigned
ATC code: not yet assigned
Mechanism of action
Avacopan is a selective antagonist of the human complement 5a receptor (C5aR1 or CD88) and competitively inhibits the interaction between C5aR1 and the anaphylatoxin C5a.
The specific and selective blockade of C5aR1 by avacopan reduces the pro-inflammatory effects of C5a, which include neutrophil activation, migration, and adherence to sites of small blood vessel inflammation, vascular endothelial cell retraction and permeability.
Pharmacodynamic effects
Avacopan blocks the C5a-induced upregulation of CD11b (integrin alpha M) on neutrophils taken from humans dosed with avacopan. CD11b facilitates neutrophil adherence to vascular endothelial surfaces, one of the steps in the vasculitis disease process.
Clinical efficacy and safety
A total of 330 patients aged 13 years or older with granulomatosis with polyangiitis (GPA) (54.8%) or microscopic polyangiitis (MPA) (45.2%) were treated in the active-comparator, randomised, doubleblind, double-dummy, multicentre, pivotal phase 3 ADVOCATE study for 52 weeks.
The ADVOCATE study design is depicted in Figure 1.
Figure 1. ADVOCATE study design:
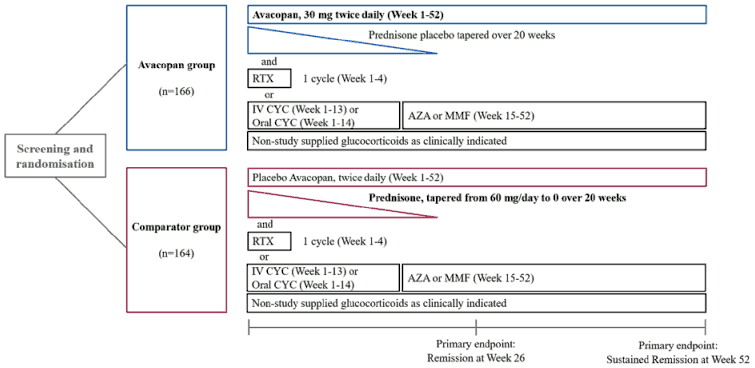
AZA = azathioprine; CYC = cyclophosphamide; IV = intravenous; MMF = mycophenolate mofetil; RTX =rituximab
Patients were randomised in a 1:1 ratio to one of the two groups:
- Avacopan group (N=166): Patients received 30 mg avacopan twice daily for 52 weeks plus prednisone-matching placebo tapering regimen over 20 weeks,
- Comparator group (N=164): Patients received avacopan-matching placebo twice daily for 52 weeks plus prednisone (tapered from 60 mg/day to 0 over 20 weeks).
All patients in both groups received standard immunosuppressive regimens of either:
- Rituximab at the dose of 375 mg/m² for 4 weekly intravenous doses, or
- Intravenous cyclophosphamide for 13 weeks (15 mg/kg up to 1.2 g every 2 to 3 weeks), and then starting on week 15 oral azathioprine 1 mg/kg daily with titration up to 2 mg/kg daily (Mycophenolate mofetil 2 g daily was allowed in place of azathioprine. If mycophenolate mofetil was not tolerated or not available, enteric coated mycophenolate sodium could be given at a target dose of 1,440 mg/day), or
- Oral cyclophosphamide for 14 weeks (2 mg/kg daily) followed by oral azathioprine or mycophenolate mofetil/sodium starting at week 15 (same dosing regimen as intravenous cyclophosphamide).
For the first rituximab infusion, 100 mg methylprednisolone, or equivalent was given before starting the infusion with rituximab. Glucocorticoid pre-medication for the second, third, and fourth rituximab infusions was allowed.
Dose reductions or adjustments in cyclophosphamide, azathioprine, and mycophenolate were allowed to conform to standard approaches to maximize safety of these medicinal products.
The following study-supplied glucocorticoid tapering schedule was used (Table 2).
Table 2. Glucocorticoid tapering schedule - Prednisone dose (mg per day):
| Study Day | Avacopan | Comparator | |
|---|---|---|---|
| All | ≥55 kg | <55 kg | |
| 1 to 7 | 0 | 60 | 45 |
| 8 to 14 | 0 | 45 | 45 |
| 15 to 21 | 0 | 30 | 30 |
| 22 to 42 | 0 | 25 | 25 |
| 43 to 56 | 0 | 20 | 20 |
| 57 to 70 | 0 | 15 | 15 |
| 71 to 98 | 0 | 10 | 10 |
| 99 to 140 | 0 | 5 | 5 |
| ≥141 | 0 | 0 | 0 |
Non-study supplied glucocorticoids, unless strictly necessary due to a condition requiring the use of glucocorticoids (such as adrenal insufficiency), had to be avoided as much as possible during the study. However, patients who experienced worsening or a relapse of their ANCA-associated vasculitis during the study could be treated with a limited course of glucocorticoids.
Patients were stratified at time of randomisation to obtain balance across treatment groups based on 3 factors:
- Newly-diagnosed or relapsed ANCA-associated vasculitis,
- Proteinase-3 (PR3) positive or myeloperoxidase (MPO) positive ANCA-associated vasculitis,
- Receiving either intravenous rituximab, intravenous cyclophosphamide, or oral cyclophosphamide.
The two treatment groups were well balanced regarding baseline demographics and disease characteristics of patients (Table 3).
Table 3. Selected baseline characteristics in the pivotal phase 3 ADVOCATE study (Intentto-Treat Population):
| Demographic characteristic | Avacopan (N = 166) | Comparator (N = 164) |
|---|---|---|
| Age at screening | ||
| Mean (SD), years | 61 (14.6) | 61 (14.5) |
| Range, years | 13-83 | 15-88 |
| ANCA-associated vasculitis status, n (%) | ||
| Newly diagnosed | 115 (69.3) | 114 (69.5) |
| Relapsed | 51 (30.7) | 50 (30.5) |
| ANCA positivity, n (%) | ||
| PR3 | 72 (43.4) | 70 (42.7) |
| MPO | 94 (56.6) | 94 (57.3) |
| Type of ANCA-associated vasculitis, n (%) | ||
| Granulomatosis with polyangiitis (GPA) | 91 (54.8) | 90 (54.9) |
| Microscopic polyangiitis (MPA) | 75 (45.2) | 74 (45.1) |
| BVAS score | ||
| Mean (SD) | 16.3 (5.87) | 16.2 (5.69) |
| eGFR | ||
| Mean (SD), mL/min/1.73 m² | 50.7 (30.96) | 52.9 (32.67) |
| Prior Glucocorticoid Use (during Screening) | ||
| n (%) | 125 (75.3) | 135 (82.3) |
| Mean (SD), prednisone-equivalent dose (mg) | 654 (744.4) | 728 (787.8) |
ANCA = antineutrophil cytoplasmic autoantibody; BVAS = Birmingham Vasculitis Activity Score;
MPO = myeloperoxidase; PR3 = proteinase-3, SD = standard deviation
The aim of the study was to determine if avacopan could provide an effective treatment for patients with ANCA-associated vasculitis, while also allowing for the reduction of glucocorticoids use without compromising safety or efficacy.
The primary objective was to evaluate the efficacy of the above described treatment regimens to induce and sustain remission in patients with ANCA-associated vasculitis based on the following two primary endpoints:
- the proportion of patients in disease remission defined as achieving a Birmingham Vasculitis Activity Score (BVAS) of 0 and not taking glucocorticoids for treatment of ANCA-associated vasculitis within 4 weeks prior to week 26,
- the proportion of patients in sustained remission defined as remission at week 26 without relapse to week 52, and BVAS of 0 and not taking glucocorticoids for treatment of ANCA-associated vasculitis within 4 weeks prior to week 52.
The two primary endpoints were tested sequentially for non-inferiority and superiority using a gatekeeping procedure to preserve the Type I error rate at 0.05.
Results from this study are showed in Table 4.
Table 4. Remission at week 26 and sustained remission at week 52 in the pivotal phase 3 ADVOCATE study (Intent-to-Treat Population):
| Avacopan N=166 n (%) | Comparator N=164 n (%) | Estimate of Treatment Difference in %a | |
|---|---|---|---|
| Remission at week 26 | 120 (72.3) | 115 (70.1) | 3.4 |
| 95% CI | 64.8, 78.9 | 62.5, 77.0 | -6.0, 12.8 |
| Sustained remission at week 52 | 109 (65.7) | 90 (54.9) | 12.5b |
| 95% CI | 57.9, 72.8 | 46.9, 62.6 | 2.6, 22.3 |
CI = confidence interval
a Two-sided 95% CIs are calculated by adjusting for randomisation stratification factors.
b superiority p value = 0.013 (2-sided)
The efficacy observed was consistent across pertinent subgroups, i.e., those with newly-diagnosed and relapsed disease, PR3 and MPO ANCA positive, GPA and MPA, and men and women. Efficacy results by background treatment are presented in Table 5.
Table 5. Remission at week 26 and sustained remission at week 52 in the pivotal phase 3 ADVOCATE study by background treatment (Intent-to-Treat Population):
| Avacopan n/N (%) | Comparator n/N (%) | Difference in %, 95% CIa | |
|---|---|---|---|
| Remission at week 26 | |||
| Patients receiving intravenous rituximab | 83/107 (77.6) | 81/107 (75.7) | 1.9 (-9.5, 13.2) |
| Patients receiving intravenous or oral cyclophosphamide | 37/59 (62.7) | 34/57 (59.6) | 3.1 (-14.7, 20.8) |
| Sustained remission at week 52 | |||
| Patients receiving intravenous rituximab | 76/107 (71.0) | 60/107 (56.1) | 15.0 (2.2, 27.7) |
| Patients receiving intravenous or oral cyclophosphamide | 33/59 (55.9) | 30/57 (52.6) | 3.3 (-14.8, 21.4) |
a Two-sided 95% confidence intervals (CI) are calculated for the difference in proportions (avacopan minus comparator) using the Wald method.
Glucocorticoid toxicity
In the pivotal phase 3 ADVOCATE study, the mean total cumulative prednisone-equivalent dose from day 1 to end-of-treatment was approximately 2.7-fold higher in the comparator group versus the avacopan group (3654.5 mg vs 1348.9 mg, respectively).
From baseline to week 26, 86.1% of patients using avacopan received non-study supplied glucocorticoids. In the comparator group, the majority of glucocorticoids use was due to the protocoldefined prednisone course.
Figure 2. Total mean daily prednisone-equivalent glucocorticoid dose per patient by study week in the ADVOCATE study (Intent-to-Treat Population):
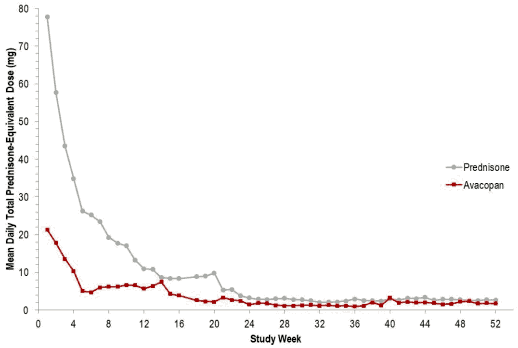
The Glucocorticoid Toxicity Index (GTI) assesses glucocorticoid-related morbidity, including measures of body mass index, glucose tolerance, lipids, steroid myopathy, skin toxicity, neuropsychiatric toxicity, and infection. A higher GTI indicates greater glucocorticoid toxicity. The GTI contains the Cumulative Worsening Score (CWS) that captures cumulative toxicity over the course of time, and the Aggregate Improvement Score (AIS) that captures both improvement and worsening of toxicity over time.
The two GTI scores (CWS and AIS) of the avacopan group versus the comparator group are summarised in Table 6. The GTI measures were secondary endpoints in the study and not controlled for multiplicity
Table 6. Glucocorticoid Toxicity Index results in the pivotal phase 3 ADVOCATE study (Intent-to-Treat Population):
| Avacopan N = 166 | Comparator N = 164 | Difference between Groups, 95% CI | |
|---|---|---|---|
| Cumulative Worsening Score (CWS) | |||
| Week 13 (least squares mean) | 25.7 | 36.6 | -11.0 (-19.7, -2.2) |
| Week 26 (least squares mean) | 39.7 | 56.6 | -16.8 (-25.6, -8.0) |
| Aggregate Improvement Score (AIS) | |||
| Week 13 (least squares mean) | 9.9 | 23.2 | -13.3 (-22.2, -4.4) |
| Week 26 (least squares mean) | 11.2 | 23.4 | -12.1 (-21.1, -3.2) |
Paediatric population
A total of 3 adolescents were studied in the pivotal phase 3 ADVOCATE study, two in the avacopan group and one in the comparator group. One adolescent in the avacopan group discontinued treatment due to worsening renal vasculitis. The second adolescent patient who received avacopan completed treatment, achieved both remission at week 26 and sustained remission at week 52.
The adolescent in the comparator group discontinued treatment due to non-adherence to contraception.
The European Medicines Agency has deferred the obligation to submit the results of studies with avacopan in one or more subsets of the paediatric population in treatment of ANCA-associated vasculitis (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
When administered without food, avacopan peak plasma concentration (Cmax) occurs at a median time (tmax) of approximately 2 hours. Avacopan has shown an approximate dose-proportional increase in systemic exposure in the dose range of 10 to 30 mg.
Co-administration of 30 mg in capsule formulation with a high-fat, high-calorie meal increases the plasma exposure (AUC) of avacopan by approximately 72% and delays tmax by approximately 3 hours; however, the Cmax is not affected.
Distribution
The reversible plasma protein binding (e.g., to albumin and α1-acid glycoprotein) of avacopan and metabolite M1 is greater than 99.9%. The apparent volume of distribution is high (Vz/F 3,000-11,000 L), indicating broad tissue distribution of the active substance.
Biotransformation
Avacopan is eliminated mainly through phase I metabolism. Following oral administration of radiolabelled avacopan, the bulk of the active substance-related materials was recovered in faeces in the form of phase I metabolites. One major circulating metabolite (M1), a mono-hydroxylated product of avacopan, was present at ~12% of the total active substance-related materials in plasma. This metabolite constitutes 30 to 50% of the parent exposure and has approximately the same activity as avacopan on C5aR1. Cytochrome P450 (CYP) 3A4 is the major enzyme responsible for the clearance of avacopan and for the formation and clearance of metabolite M1.
Avacopan is a weak inhibitor of CYP3A4 and CYP2C9 as indicated by a modest increase in the AUC of the probe active substances midazolam (1.81-fold) and celecoxib (1.15-fold), respectively.
In vitro, avacopan is not an inhibitor or an inducer of other CYP enzymes.
Avacopan showed negligible to weak inhibition of common transporters in vitro. Therefore, clinically relevant interactions are unlikely when avacopan is co-administered with substances that are substrates or inhibitors of these transporters.
Elimination
Based on population pharmacokinetic analysis, the total apparent body clearance (CL/F) of avacopan is 16.3 L/h (95% CI: 13.1-21.1 L/h). The median terminal elimination half-life is 510 hours (21 days) based on population pharmacokinetic analysis. When avacopan is stopped after steady state has been reached, the residual plasma concentration of avacopan is projected to decrease to ~20%, <10%, and <5% of the steady state maximum concentration approximately 4 weeks, 7 weeks, and 10 weeks, respectively, after the last dose.
Following oral administration of radiolabelled avacopan, about 77% and 10% of the radioactivity was recovered in faeces and urine, respectively, and 7% and <0.1% of the radioactive dose was recovered as unchanged avacopan in faeces and urine, respectively. These results suggest that the main route of clearance of avacopan is metabolism followed by biliary excretion of the metabolites into faeces, and that direct excretion of avacopan into urine or faeces via bile is negligible.
Special populations
Elderly
Population pharmacokinetic analysis found no significant effect of age (among adults) on the plasma exposure of avacopan; however, there were limited pharmacokinetic data in patients over 75 years of age in clinical studies. No dose adjustment is necessary for elderly patients (see section 4.2).
Hepatic impairment
The pharmacokinetic properties of avacopan have been examined in 16 subjects with mild (ChildPugh class A) or moderate (Child-Pugh class B) hepatic impairment. When compared to normal controls, no pharmacologically relevant differences in exposure (mean ratios of Cmax and AUC ≤1.3) of avacopan or its major metabolite M1 was observed; therefore, no dose adjustment is necessary (see section 4.2).
Avacopan has not been studied in subjects with severe hepatic impairment (Child-Pugh class C) (see section 4.2).
Renal impairment
Based on population pharmacokinetic analysis, the plasma exposure of avacopan is similar between patients with renal impairment and healthy subjects. Therefore, no dose adjustment is necessary based on renal function (see section 4.2).
Avacopan has not been studied in patients with ANCA-associated vasculitis with an eGFR below 15 mL/min/1.73 m², who are on dialysis, in need of dialysis or plasma exchange.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity and carcinogenicity.
Fertility and early embryonic development
Avacopan produced no effects on male or female reproductive performance (fertility) or early development in hamsters at oral doses equivalent up to 6.8-fold the clinical AUC.
Embryo-foetal development
Avacopan was not teratogenic when dosed orally to hamsters and rabbits. In hamsters, an increased incidence of skeletal variations (short thoracolumbar supernumerary rib) was observed at exposure equivalent to 5.3-fold the clinical AUC. In rabbits, avacopan caused maternal toxicity (adverse clinical signs and abortions), but no foetal toxicity at 0.6-fold the clinical AUC.
Pre- and post-natal development
Avacopan did not result in adverse effects in female offspring when administered in hamsters at exposures up to 6.3-fold the clinical AUC during gestation and through lactation until weaning. In males, there was a slight delay in preputial separation at 3.7-fold the clinical AUC. This isolated finding was considered to be of low toxicological significance and was not associated with any impairment of reproductive performance.
Analysis of avacopan plasma levels in the lactating dams and the plasma levels in nursing offspring showed the presence of avacopan, suggesting that avacopan is likely secreted into the milk of lactating hamsters.
Carcinogenicity
The carcinogenic potential of avacopan was evaluated in a 2-year study in both rats and hamsters. In male rats, a slightly increased incidence of C-cell thyroid adenoma was noted in avacopan-treated rats; this increase was not statistically significant, and the incidence was within the historical control range. Avacopan was not carcinogenic in hamsters, the pharmacologically relevant species.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.