TRIJARDY XR Extended-release tablet Ref.[50504] Active ingredients: Empagliflozin Linagliptin Metformin
Source: FDA, National Drug Code (US) Revision Year: 2022
12.1. Mechanism of Action
TRIJARDY XR
TRIJARDY XR contains: empagliflozin, a sodium-glucose co-transporter 2 (SGLT2) inhibitor, linagliptin, a dipeptidyl peptidase-4 (DPP-4) inhibitor, and metformin, a biguanide.
Empagliflozin
Empagliflozin is an inhibitor of the sodium-glucose co-transporter 2 (SGLT2), the predominant transporter responsible for reabsorption of glucose from the glomerular filtrate back into the circulation. By inhibiting SGLT2, empagliflozin reduces renal reabsorption of filtered glucose and lowers the renal threshold for glucose, and thereby increases urinary glucose excretion.
Linagliptin
Linagliptin is an inhibitor of DPP-4, an enzyme that degrades the incretin hormones glucagon-like peptide-1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP). Thus, linagliptin increases the concentrations of active incretin hormones, stimulating the release of insulin in a glucose-dependent manner and decreasing the levels of glucagon in the circulation. Both incretin hormones are involved in the physiological regulation of glucose homeostasis. Incretin hormones are secreted at a low basal level throughout the day and levels rise immediately after meal intake. GLP-1 and GIP increase insulin biosynthesis and secretion from pancreatic beta cells in the presence of normal and elevated blood glucose levels. Furthermore, GLP-1 also reduces glucagon secretion from pancreatic alpha cells, resulting in a reduction in hepatic glucose output.
Metformin HCl
Metformin is an antihyperglycemic agent which improves glucose tolerance in patients with type 2 diabetes mellitus, lowering both basal and postprandial plasma glucose. Metformin decreases hepatic glucose production, decreases intestinal absorption of glucose, and improves insulin sensitivity by increasing peripheral glucose uptake and utilization. With metformin therapy, insulin secretion remains unchanged while fasting insulin levels and day-long plasma insulin response may decrease.
12.2. Pharmacodynamics
Empagliflozin
Urinary Glucose Excretion
In patients with type 2 diabetes, urinary glucose excretion increased immediately following a dose of empagliflozin and was maintained at the end of a 4-week treatment period averaging at approximately 64 grams per day with 10 mg empagliflozin and 78 grams per day with 25 mg empagliflozin once daily. Data from single oral doses of empagliflozin in healthy subjects indicate that, on average, the elevation in urinary glucose excretion approaches baseline by about 3 days for the 10 mg and 25 mg doses.
Urinary Volume
In a 5-day study, mean 24-hour urine volume increase from baseline was 341 mL on Day 1 and 135 mL on Day 5 of empagliflozin 25 mg once daily treatment.
Cardiac Electrophysiology
In a randomized, placebo-controlled, active-comparator, crossover study, 30 healthy subjects were administered a single oral dose of empagliflozin 25 mg, empagliflozin 200 mg (8 times the maximum recommended dose), moxifloxacin, and placebo. No increase in QTc was observed with either 25 mg or 200 mg empagliflozin.
Linagliptin
Linagliptin binds to DPP-4 in a reversible manner and increases the concentrations of incretin hormones. Linagliptin glucose-dependently increases insulin secretion and lowers glucagon secretion, thus resulting in a better regulation of the glucose homeostasis. Linagliptin binds selectively to DPP-4 and selectively inhibits DPP-4, but not DPP-8 or DPP-9 activity in vitro at concentrations approximating therapeutic exposures.
Cardiac Electrophysiology
In a randomized, placebo-controlled, active-comparator, 4-way crossover study, 36 healthy subjects were administered a single oral dose of linagliptin 5 mg, linagliptin 100 mg (20 times the recommended dose), moxifloxacin, and placebo. No increase in QTc was observed with either the recommended dose of 5 mg or the 100-mg dose. At the 100-mg dose, peak linagliptin plasma concentrations were approximately 38-fold higher than the peak concentrations following a 5-mg dose.
12.3. Pharmacokinetics
Administration of TRIJARDY XR with food resulted in no change in overall exposure of empagliflozin or linagliptin. For metformin extended-release, high-fat meals increased systemic exposure (as measured by area-under-the-curve [AUC]) by approximately 70% relative to fasting, while Cmax is not affected. Meals prolonged Tmax by approximately 3 hours.
Empagliflozin
Absorption
The pharmacokinetics of empagliflozin has been characterized in healthy volunteers and patients with type 2 diabetes and no clinically relevant differences were noted between the two populations. After oral administration, peak plasma concentrations of empagliflozin were reached at 1.5 hours post-dose. Thereafter, plasma concentrations declined in a biphasic manner with a rapid distribution phase and a relatively slow terminal phase. The steady-state mean plasma AUC and Cmax were 1870 nmol∙h/L and 259 nmol/L, respectively, with 10 mg empagliflozin once daily treatment, and 4740 nmol∙h/L and 687 nmol/L, respectively, with 25 mg empagliflozin once daily treatment. Systemic exposure of empagliflozin increased in a dose-proportional manner in the therapeutic dose range. The single-dose and steady-state pharmacokinetic parameters of empagliflozin were similar, suggesting linear pharmacokinetics with respect to time.
Distribution
The apparent steady-state volume of distribution was estimated to be 73.8 L based on a population pharmacokinetic analysis. Following administration of an oral [14C]-empagliflozin solution to healthy subjects, the red blood cell partitioning was approximately 36.8% and plasma protein binding was 86.2%.
Elimination
The apparent terminal elimination half-life of empagliflozin was estimated to be 12.4 h and apparent oral clearance was 10.6 L/h based on the population pharmacokinetic analysis. Following once-daily dosing, up to 22% accumulation, with respect to plasma AUC, was observed at steady-state, which was consistent with empagliflozin half-life.
Metabolism
No major metabolites of empagliflozin were detected in human plasma and the most abundant metabolites were three glucuronide conjugates (2-O-, 3-O-, and 6-O-glucuronide). Systemic exposure of each metabolite was less than 10% of total drug-related material. In vitro studies suggested that the primary route of metabolism of empagliflozin in humans is glucuronidation by the uridine 5'-diphospho-glucuronosyltransferases UGT2B7, UGT1A3, UGT1A8, and UGT1A9.
Excretion
Following administration of an oral [14C]-empagliflozin solution to healthy subjects, approximately 95.6% of the drug-related radioactivity was eliminated in feces (41.2%) or urine (54.4%). The majority of drug-related radioactivity recovered in feces was unchanged parent drug and approximately half of drug-related radioactivity excreted in urine was unchanged parent drug.
Linagliptin
Absorption
The absolute bioavailability of linagliptin is approximately 30%. A high-fat meal reduced Cmax by 15% and increased AUC by 4%; this effect is not clinically relevant. Linagliptin may be administered with or without food.
Distribution
The mean apparent volume of distribution at steady-state following a single intravenous dose of linagliptin 5 mg to healthy subjects is approximately 1110 L, indicating that linagliptin extensively distributes to the tissues. Plasma protein binding of linagliptin is concentration-dependent, decreasing from about 99% at 1 nmol/L to 75% to 89% at ≥30 nmol/L, reflecting saturation of binding to DPP-4 with increasing concentration of linagliptin. At high concentrations, where DPP-4 is fully saturated, 70% to 80% of linagliptin remains bound to plasma proteins and 20% to 30% is unbound in plasma. Plasma binding is not altered in patients with renal or hepatic impairment.
Elimination
Linagliptin has a terminal half-life of about 200 hours at steady-state, though the accumulation half-life is about 11 hours. Renal clearance at steady-state was approximately 70 mL/min.
Metabolism
Following oral administration, the majority (about 90%) of linagliptin is excreted unchanged, indicating that metabolism represents a minor elimination pathway. A small fraction of absorbed linagliptin is metabolized to a pharmacologically inactive metabolite, which shows a steady-state exposure of 13.3% relative to linagliptin.
Excretion
Following administration of an oral [ 14C]-linagliptin dose to healthy subjects, approximately 85% of the administered radioactivity was eliminated via the enterohepatic system (80%) or urine (5%) within 4 days of dosing.
Metformin HCl
Absorption
Following a single oral dose of 1000 mg (2 × 500 mg tablets) metformin HCl extended-release after a meal, the time to reach maximum plasma metformin concentration (Tmax) is achieved at approximately 7 to 8 hours. In both single- and multiple-dose studies in healthy subjects, once daily 1000 mg (2 × 500 mg tablets) dosing provides equivalent systemic exposure, as measured by AUC, and up to 35% higher Cmax of metformin relative to the immediate-release given as 500 mg twice daily.
Single oral doses of metformin HCl extended-release from 500 mg to 2500 mg resulted in less than proportional increase in both AUC and Cmax. Low-fat and high-fat meals increased the systemic exposure (as measured by AUC) from metformin extended-release tablets by about 38% and 73%, respectively, relative to fasting. Both meals prolonged metformin Tmax by approximately 3 hours but Cmax, was not affected.
Distribution
The apparent volume of distribution (V/F) of metformin following single oral doses of immediate-release metformin HCl tablets 850 mg averaged 654±358 L. Metformin is negligibly bound to plasma proteins. Metformin partitions into erythrocytes, most likely as a function of time.
Elimination
Metformin has a plasma elimination half-life of approximately 6.2 hours. In blood, the elimination half-life is approximately 17.6 hours, suggesting that the erythrocyte mass may be a compartment of distribution.
Metabolism
Intravenous single-dose studies in normal subjects demonstrate that metformin does not undergo hepatic metabolism (no metabolites have been identified in humans) nor biliary excretion.
Excretion
Following oral administration, approximately 90% of the absorbed drug is excreted via the renal route within the first 24 hours. Renal clearance is approximately 3.5 times greater than creatinine clearance, which indicates that tubular secretion is the major route of metformin elimination.
Specific Populations
Renal Impairment
TRIJARDY XR
Studies characterizing the pharmacokinetics of empagliflozin, linagliptin, and metformin after administration of TRIJARDY XR in renally impaired patients have not been performed.
Empagliflozin
In patients with mild (eGFR: 60 to less than 90 mL/min/1.73 m 2), moderate (eGFR: 30 to less than 60 mL/min/1.73 m 2), and severe (eGFR: less than 30 mL/min/1.73 m 2) renal impairment and patients on dialysis due to kidney failure, AUC of empagliflozin increased by approximately 18%, 20%, 66%, and 48%, respectively, compared to subjects with normal renal function. Peak plasma levels of empagliflozin were similar in patients with moderate renal impairment and patients on dialysis due to kidney failure compared to subjects with normal renal function. Peak plasma levels of empagliflozin were roughly 20% higher in patients with mild and severe renal impairment, as compared to subjects with normal renal function. Population pharmacokinetic analysis showed that the apparent oral clearance of empagliflozin decreased, with a decrease in eGFR leading to an increase in drug exposure. However, the fraction of empagliflozin that was excreted unchanged in urine, and urinary glucose excretion, declined with decrease in eGFR.
Linagliptin
An open-label pharmacokinetic study evaluated the pharmacokinetics of linagliptin 5 mg in male and female patients with varying degrees of chronic renal impairment. The study included 6 healthy subjects with normal renal function (creatinine clearance [CrCl] ≥80 mL/min), 6 patients with mild renal impairment (CrCl 50 to <80 mL/min), 6 patients with moderate renal impairment (CrCl 30 to <50 mL/min), 10 patients with type 2 diabetes and severe renal impairment (CrCl <30 mL/min), and 11 patients with type 2 diabetes and normal renal function. Creatinine clearance was measured by 24-hour urinary creatinine clearance measurements or estimated from serum creatinine based on the Cockcroft-Gault formula.
Under steady-state conditions, linagliptin exposure in patients with mild renal impairment was comparable to healthy subjects.
In patients with moderate renal impairment under steady-state conditions, mean exposure of linagliptin increased (AUCτ,ss by 71% and Cmax by 46%), compared with healthy subjects. This increase was not associated with a prolonged accumulation half-life, terminal half-life, or an increased accumulation factor. Renal excretion of linagliptin was below 5% of the administered dose and was not affected by decreased renal function. Patients with type 2 diabetes and severe renal impairment showed steady-state exposure approximately 40% higher than that of patients with type 2 diabetes and normal renal function (increase in AUCτ,ss by 42% and Cmax by 35%). For both type 2 diabetes groups, renal excretion was below 7% of the administered dose.
These findings were further supported by the results of population pharmacokinetic analyses.
Metformin HCl
In patients with decreased renal function, the plasma and blood half-life of metformin is prolonged and the renal clearance is decreased [see Contraindications (4) and Warnings and Precautions (5.1)].
Hepatic Impairment
TRIJARDY XR
Studies characterizing the pharmacokinetics of empagliflozin, linagliptin, and metformin after administration of TRIJARDY XR in hepatically impaired patients have not been performed.
Empagliflozin
In patients with mild, moderate, and severe hepatic impairment according to the Child-Pugh classification, AUC of empagliflozin increased by approximately 23%, 47%, and 75% and Cmax increased by approximately 4%, 23%, and 48%, respectively, compared to subjects with normal hepatic function.
Linagliptin
In patients with mild hepatic impairment (Child-Pugh class A) steady-state exposure (AUCτ,ss) of linagliptin was approximately 25% lower and Cmax,ss was approximately 36% lower than in healthy subjects. In patients with moderate hepatic impairment (Child-Pugh class B), AUCss of linagliptin was about 14% lower and Cmax,ss was approximately 8% lower than in healthy subjects. Patients with severe hepatic impairment (Child-Pugh class C) had comparable exposure of linagliptin in terms of AUC0-24 and approximately 23% lower Cmax compared with healthy subjects. Reductions in the pharmacokinetic parameters seen in patients with hepatic impairment did not result in reductions in DPP-4 inhibition.
Metformin HCl
No pharmacokinetic studies of metformin have been conducted in patients with hepatic impairment.
Effects of Age, Body Mass Index, Gender, and Race
Empagliflozin
Based on the population PK analysis, age, body mass index (BMI), gender and race (Asians versus primarily Whites) do not have a clinically meaningful effect on pharmacokinetics of empagliflozin [see Use in Specific Populations (8.5)].
Linagliptin
Based on the population PK analysis, age, body mass index (BMI), gender and race do not have a clinically meaningful effect on pharmacokinetics of linagliptin [see Use in Specific Populations (8.5)].
Metformin HCl
Metformin pharmacokinetic parameters did not differ significantly between normal subjects and patients with type 2 diabetes mellitus when analyzed according to gender. Similarly, in controlled clinical studies in patients with type 2 diabetes mellitus, the antihyperglycemic effect of metformin was comparable in males and females.
No studies of metformin pharmacokinetic parameters according to race have been performed. In controlled clinical studies of metformin HCl in patients with type 2 diabetes mellitus, the antihyperglycemic effect was comparable in Caucasians (n=249), Blacks (n=51), and Hispanics (n=24).
Limited data from controlled pharmacokinetic studies of metformin HCl in healthy elderly subjects suggest that total plasma clearance of metformin is decreased, the half-life is prolonged, and Cmax is increased, compared with healthy young subjects. From these data, it appears that the change in metformin pharmacokinetics with aging is primarily accounted for by a change in renal function.
Drug Interactions
Pharmacokinetic drug interaction studies with TRIJARDY XR have not been performed; however, such studies have been conducted with the individual components of TRIJARDY XR (empagliflozin, linagliptin, and metformin HCl).
Empagliflozin
In vitro Assessment of Drug Interactions
Empagliflozin does not inhibit, inactivate, or induce CYP450 isoforms. In vitro data suggest that the primary route of metabolism of empagliflozin in humans is glucuronidation by the uridine 5'-diphospho-glucuronosyltransferases UGT1A3, UGT1A8, UGT1A9 and UGT2B7. Empagliflozin does not inhibit UGT1A1, UGT1A3, UGT1A8, UGT1A9, or UGT2B7. Therefore, no effect of empagliflozin is anticipated on concomitantly administered drugs that are substrates of the major CYP450 isoforms or UGT1A1, UGT1A3, UGT1A8, UGT1A9, or UGT2B7. The effect of UGT induction (e.g., induction by rifampicin or any other UGT enzyme inducer) on empagliflozin exposure has not been evaluated.
Empagliflozin is a substrate for P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP), but it does not inhibit these efflux transporters at therapeutic doses. Based on in vitro studies, empagliflozin is considered unlikely to cause interactions with drugs that are P-gp substrates. Empagliflozin is a substrate of the human uptake transporters OAT3, OATP1B1, and OATP1B3, but not OAT1 and OCT2. Empagliflozin does not inhibit any of these human uptake transporters at clinically relevant plasma concentrations and, therefore, no effect of empagliflozin is anticipated on concomitantly administered drugs that are substrates of these uptake transporters.
In vivo Assessment of Drug Interactions
Empagliflozin pharmacokinetics were similar with and without coadministration of metformin, glimepiride, pioglitazone, sitagliptin, linagliptin, warfarin, verapamil, ramipril, and simvastatin in healthy volunteers and with or without coadministration of hydrochlorothiazide and torsemide in patients with type 2 diabetes (see Figure 1). In subjects with normal renal function, coadministration of empagliflozin with probenecid resulted in a 30% decrease in the fraction of empagliflozin excreted in urine without any effect on 24-hour urinary glucose excretion. The relevance of this observation to patients with renal impairment is unknown.
Figure 1. Effect of Various Medications on the Pharmacokinetics of Empagliflozin as Displayed as 90% Confidence Interval of Geometric Mean AUC and Cmax Ratios [reference lines indicate 100% (80% - 125%)]:
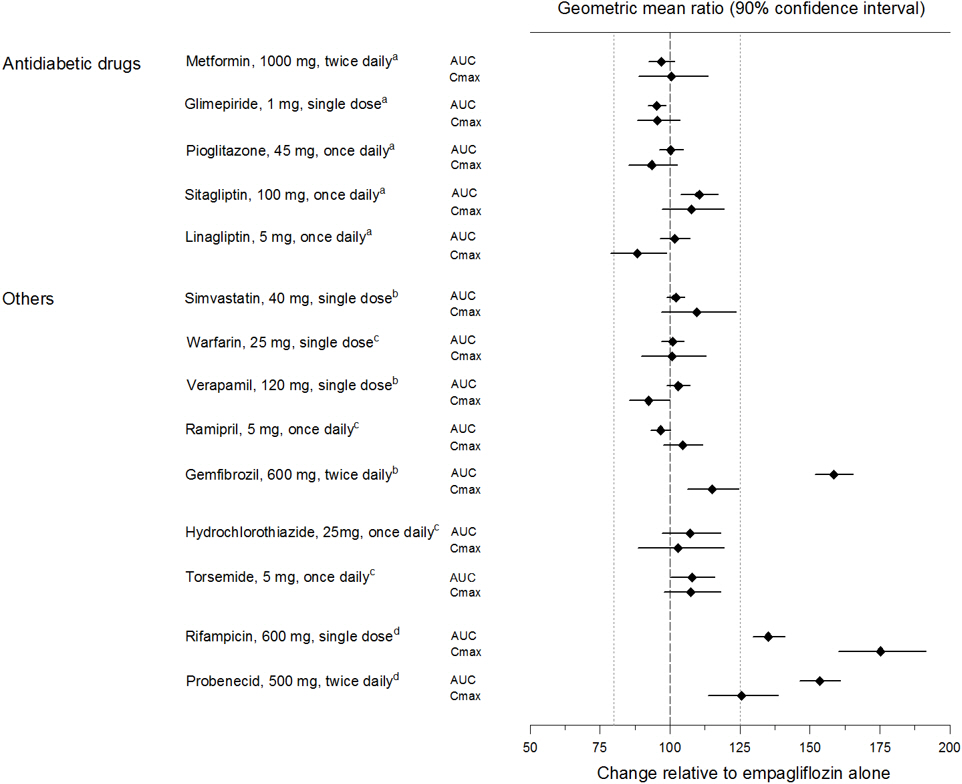
a empagliflozin, 50 mg, once daily
b empagliflozin, 25 mg, single dose
c empagliflozin, 25 mg, once daily
d empagliflozin, 10 mg, single dose
Empagliflozin had no clinically relevant effect on the pharmacokinetics of metformin, glimepiride, pioglitazone, sitagliptin, linagliptin, warfarin, digoxin, ramipril, simvastatin, hydrochlorothiazide, torsemide, and oral contraceptives when coadministered in healthy volunteers (see Figure 2).
Figure 2. Effect of Empagliflozin on the Pharmacokinetics of Various Medications as Displayed as 90% Confidence Interval of Geometric Mean AUC and Cmax Ratios [reference lines indicate 100% (80% - 125%)]:
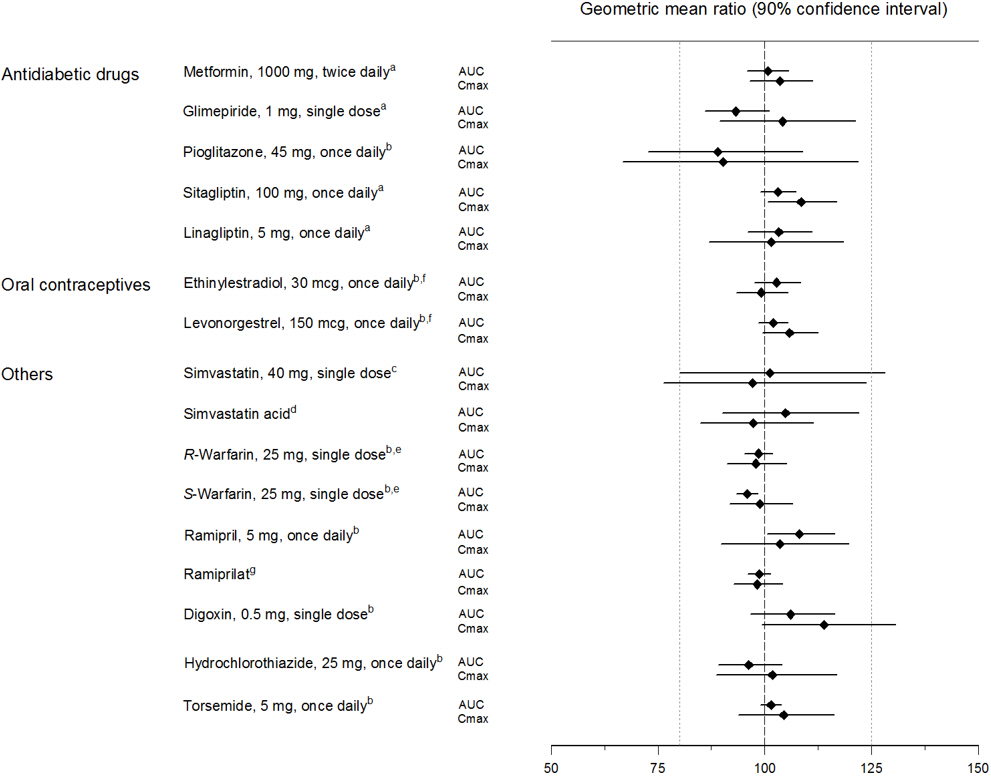
a empagliflozin, 50 mg, once daily
b empagliflozin, 25 mg, once daily
c empagliflozin, 25 mg, single dose
d administered as simvastatin
e administered as warfarin racemic mixture
f administered as Microgynon
g administered as ramipril
Linagliptin
In vitro Assessment of Drug Interactions
Linagliptin is a weak to moderate inhibitor of CYP isozyme CYP3A4 but does not inhibit other CYP isozymes and is not an inducer of CYP isozymes, including CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 4A11.
Linagliptin is a P-glycoprotein (P-gp) substrate and inhibits P-gp mediated transport of digoxin at high concentrations. Based on these results, and in vivo drug interaction studies, linagliptin is considered unlikely to cause interactions with other P-gp substrates at therapeutic concentrations.
In vivo Assessment of Drug Interactions
Strong inducers of CYP3A4 or P-gp (e.g., rifampin) decrease exposure to linagliptin to subtherapeutic and likely ineffective concentrations [see Drug Interactions (7)]. In vivo studies indicated evidence of a low propensity for causing drug interactions with substrates of CYP3A4, CYP2C9, CYP2C8, P-gp and organic cationic transporter (OCT).
Table 3. Effect of Coadministered Drugs on Systemic Exposure of Linagliptin:
| Coadministered Drug | Dosing of Coadministered Druga | Dosing of Linagliptina | Geometric Mean Ratio (ratio with/without coadministered drug) No effect=1.0 | |
|---|---|---|---|---|
| AUCd | Cmax | |||
| Metformin | 850 mg TID | 10 mg QD | 1.20 | 1.03 |
| Glyburide | 1.75 mg c | 5 mg QD | 1.02 | 1.01 |
| Pioglitazone | 45 mg QD | 10 mg QD | 1.13 | 1.07 |
| Ritonavir | 200 mg BID | 5 mg c | 2.01 | 2.96 |
| Rifampinb | 600 mg QD | 5 mg QD | 0.60 | 0.56 |
a Multiple dose (steady-state) unless otherwise noted
b For information regarding clinical recommendations [see Drug Interactions (7)]
c Single dose
d AUC = AUC(0 to 24 hours) for single dose treatments and AUC = AUC(TAU) for multiple-dose treatments
QD = once daily
BID = twice daily
TID = three times daily
Table 4. Effect of Linagliptin on Systemic Exposure of Coadministered Drugs:
| Coadministered Drug | Dosing of Coadministered Druga | Dosing of Linagliptina | Geometric Mean Ratio (ratio with/without coadministered drug) No effect=1.0 | ||
|---|---|---|---|---|---|
| AUCc | Cmax | ||||
| Metformin | 850 mg TID | 10 mg QD | metformin | 1.01 | 0.89 |
| Glyburide | 1.75 mg b | 5 mg QD | glyburide | 0.86 | 0.86 |
| Pioglitazone | 45 mg QD | 10 mg QD | pioglitazone | 0.94 | 0.86 |
| metabolite M-III | 0.98 | 0.96 | |||
| metabolite M-IV | 1.04 | 1.05 | |||
| Digoxin | 0.25 mg QD | 5 mg QD | digoxin | 1.02 | 0.94 |
| Simvastatin | 40 mg QD | 10 mg QD | simvastatin | 1.34 | 1.10 |
| simvastatin acid | 1.33 | 1.21 | |||
| Warfarin | 10 mg b | 5 mg QD | R-warfarin | 0.99 | 1.00 |
| S-warfarin | 1.03 | 1.01 | |||
| INR | 0.93 d | 1.04 d | |||
| PT | 1.03 d | 1.15 d | |||
| Ethinylestradiol and levonorgestrel | ethinylestradiol 0.03 mg and levonorgestrel 0.150 mg QD | 5 mg QD | ethinylestradiol levonorgestrel | 1.01 1.09 | 1.08 1.13 |
a Multiple dose (steady-state) unless otherwise noted
b Single dose
c AUC = AUC(INF) for single dose treatments and AUC = AUC(TAU) for multiple-dose treatments
d AUC = AUC(0-168) and Cmax = Emax for pharmacodynamic end points
INR = International Normalized Ratio
PT = Prothrombin Time
QD = once daily
TID = three times daily
Metformin HCl
Table 5. Effect of Coadministered Drug on Plasma Metformin Systemic Exposure:
| Coadministered Drug | Dosing of Coadministered Drug* | Dosing of Metformin HCl* | Geometric Mean Ratio (ratio with/without coadministered drug) No effect=1.0 | ||
|---|---|---|---|---|---|
| AUC† | Cmax | ||||
| Glyburide | 5 mg | 500 mg≠ | metformin | 0.98‡ | 0.99‡ |
| Furosemide | 40 mg | 850 mg | metformin | 1.09‡ | 1.22‡ |
| Nifedipine | 10 mg | 850 mg | metformin | 1.16 | 1.21 |
| Propranolol | 40 mg | 850 mg | metformin | 0.90 | 0.94 |
| Ibuprofen | 400 mg | 850 mg | metformin | 1.05‡ | 1.07‡ |
| Cationic drugs eliminated by renal tubular secretion may reduce metformin elimination [see Drug Interactions (7)] | |||||
| Cimetidine | 400 mg | 850 mg | metformin | 1.40 | 1.61 |
| Carbonic anhydrase inhibitors may cause metabolic acidosis [see Drug Interactions (7)] | |||||
| Topiramate** | 100 mg | 500 mg | metformin | 1.25 | 1.17 |
* All metformin and coadministered drugs were given as single doses
† AUC = AUC(INF)
≠ Metformin HCl extended-release tablets 500 mg
‡ Ratio of arithmetic means
** At steady-state with topiramate 100 mg every 12 hours and metformin 500 mg every 12 hours; AUC = AUC(0-12 hours)
Table 6. Effect of Metformin on Coadministered Drug Systemic Exposure:
| Coadministered Drug | Dosing of Coadministered Drug* | Dosing of Metformin HCl* | Geometric Mean Ratio (ratio with/without metformin) No effect=1.0 | ||
|---|---|---|---|---|---|
| AUC† | Cmax | ||||
| Glyburide | 5 mg | 500 mg§ | glyburide | 0.78‡ | 0.63‡ |
| Furosemide | 40 mg | 850 mg | furosemide | 0.87‡ | 0.69‡ |
| Nifedipine | 10 mg | 850 mg | nifedipine | 1.10§ | 1.08 |
| Propranolol | 40 mg | 850 mg | propranolol | 1.01§ | 0.94 |
| Ibuprofen | 400 mg | 850 mg | ibuprofen | 0.97¶ | 1.01¶ |
| Cimetidine | 400 mg | 850 mg | cimetidine | 0.95§ | 1.01 |
* All metformin and coadministered drugs were given as single doses
† AUC = AUC(INF) unless otherwise noted
§ AUC(0-24 hours) reported
‡ Ratio of arithmetic means, p-value of difference <0.05
¶ Ratio of arithmetic means
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
TRIJARDY XR
No carcinogenicity, mutagenicity, or impairment of fertility studies have been conducted with the combination of empagliflozin, linagliptin, and metformin HCl.
Empagliflozin
Carcinogenesis was evaluated in 2-year studies conducted in CD-1 mice and Wistar rats. Empagliflozin did not increase the incidence of tumors in female rats dosed at 100, 300, or 700 mg/kg/day (up to 72 times the exposure from the maximum clinical dose of 25 mg). In male rats, hemangiomas of the mesenteric lymph node were increased significantly at 700 mg/kg/day or approximately 42 times the exposure from a 25 mg clinical dose. Empagliflozin did not increase the incidence of tumors in female mice dosed at 100, 300, or 1000 mg/kg/day (up to 62 times the exposure from a 25 mg clinical dose). Renal tubule adenomas and carcinomas were observed in male mice at 1000 mg/kg/day, which is approximately 45 times the exposure of the maximum clinical dose of 25 mg. These tumors may be associated with a metabolic pathway predominantly present in the male mouse kidney.
Empagliflozin was not mutagenic or clastogenic with or without metabolic activation in the in vitro Ames bacterial mutagenicity assay, the in vitro L5178Y tk+/- mouse lymphoma cell assay, and an in vivo micronucleus assay in rats.
Empagliflozin had no effects on mating, fertility or early embryonic development in treated male or female rats, up to the high dose of 700 mg/kg/day (approximately 155 times the 25 mg clinical dose in males and females, respectively).
Linagliptin
Linagliptin did not increase the incidence of tumors in male and female rats in a 2-year study at doses of 6, 18, and 60 mg/kg. The highest dose of 60 mg/kg is approximately 418 times the clinical dose of 5 mg/day based on AUC exposure. Linagliptin did not increase the incidence of tumors in mice in a 2-year study at doses up to 80 mg/kg (males) and 25 mg/kg (females), or approximately 35 and 270 times the clinical dose based on AUC exposure. Higher doses of linagliptin in female mice (80 mg/kg) increased the incidence of lymphoma at approximately 215 times the clinical dose based on AUC exposure.
Linagliptin was not mutagenic or clastogenic with or without metabolic activation in the Ames bacterial mutagenicity assay, a chromosomal aberration test in human lymphocytes, and an in vivo micronucleus assay.
In fertility studies in rats, linagliptin had no adverse effects on early embryonic development, mating, fertility, or bearing live young up to the highest dose of 240 mg/kg (approximately 943 times the clinical dose based on AUC exposure).
Metformin HCl
Long-term carcinogenicity studies have been performed in Sprague Dawley rats at doses of 150, 300, and 450 mg/kg/day in males and 150, 450, 900, and 1200 mg/kg/day in females. These doses are approximately 2, 4, and 8 times in males, and 3, 7, 12, and 16 times in females of the maximum recommended human daily dose of 2000 mg/kg/day based on body surface area comparisons. No evidence of carcinogenicity with metformin was found in either male or female rats. A carcinogenicity study was also performed in Tg.AC transgenic mice at doses of up to 2000 mg/kg/day applied dermally. No evidence of carcinogenicity was observed in male or female mice.
Genotoxicity assessments in the Ames test, gene mutation test (mouse lymphoma cells), chromosomal aberrations test (human lymphocytes) and in vivo mouse micronucleus tests were negative.
Fertility of male or female rats was not affected by metformin when administered at doses up to 600 mg/kg/day, which is approximately 3 times the maximum recommended human daily dose based on body surface area comparisons.
14. Clinical Studies
Empagliflozin and Linagliptin Add-on Combination Therapy with Metformin for Glycemic Control
A total of 686 patients with type 2 diabetes participated in a double-blind, active-controlled study to evaluate the efficacy and safety of empagliflozin 10 mg or 25 mg in combination with linagliptin 5 mg, compared to the individual components.
Patients with type 2 diabetes inadequately controlled on at least 1500 mg of metformin per day entered a single-blind placebo run-in period for 2 weeks. At the end of the run-in period, patients who remained inadequately controlled and had an HbA1c between 7% and 10.5% were randomized 1:1:1:1:1 to one of 5 active-treatment arms of empagliflozin 10 mg or 25 mg, linagliptin 5 mg, or linagliptin 5 mg in combination with 10 mg or 25 mg empagliflozin as a fixed dose combination tablet.
At Week 24, empagliflozin 10 mg or 25 mg used in combination with linagliptin 5 mg provided statistically significant improvement in HbA1c (p-value <0.0001) and FPG (p-value <0.001) compared to the individual components in patients who had been inadequately controlled on metformin (see Table 7, Figure 3). Treatment with empagliflozin 10 mg or 25 mg used in combination with linagliptin 5 mg also resulted in a statistically significant reduction in body weight compared to linagliptin 5 mg (p-value <0.0001). There was no statistically significant difference compared to empagliflozin alone.
Table 7. Glycemic Parameters at 24 Weeks in a Study Comparing Empagliflozin in Combination with Linagliptin to the Individual Components as Add-on Therapy in Patients Inadequately Controlled on Metformin:
| Empagliflozin 10 mg/Linagliptin 5 mg | Empagliflozin 25 mg/Linagliptin 5 mg | Empagliflozin 10 mg | Empagliflozin 25 mg | Linagliptin 5 mg | |
|---|---|---|---|---|---|
| HbA1c (%) | |||||
| Number of patients | n=135 | n=133 | n=137 | n=139 | n=128 |
| Baseline (mean) | 8.0 | 7.9 | 8.0 | 8.0 | 8.0 |
| Change from baseline (adjusted mean) | -1.1 | -1.2 | -0.7 | -0.6 | -0.7 |
| Comparison vs empagliflozin 25 mg or 10 mg (adjusted mean) (95% CI)a | -0.4 (-0.6, -0.2)d | -0.6 (-0.7, -0.4)d | -- | -- | -- |
| Comparison vs linagliptin 5 mg (adjusted mean) (95% CI)a | -0.4 (-0.6, -0.2)d | -0.5 (-0.7, -0.3)d | -- | -- | -- |
| Patients [n (%)] achieving HbA1c <7%b | 74 (58) | 76 (62) | 35 (28) | 43 (33) | 43 (36) |
| FPG (mg/dL) | |||||
| Number of patients | n=133 | n=131 | n=136 | n=137 | n=125 |
| Baseline (mean) | 157 | 155 | 162 | 160 | 156 |
| Change from baseline (adjusted mean) | -33 | -36 | -21 | -21 | -13 |
| Comparison vs empagliflozin 25 mg or 10 mg (adjusted mean) (95% CI)a | -12 (-18, -5)d | -15 (-22, -9)d | -- | -- | -- |
| Comparison vs linagliptin 5 mg (adjusted mean) (95% CI)a | -20 (-27, -13)d | -23 (-29, -16)d | -- | -- | -- |
| Body Weight | |||||
| Number of patients | n=135 | n=134 | n=137 | n=140 | n=128 |
| Baseline (mean) in kg | 87 | 85 | 86 | 88 | 85 |
| % change from baseline (adjusted mean) | -3.1 | -3.4 | -3.0 | -3.5 | -0.7 |
| Comparison vs empagliflozin 25 mg or 10 mg (adjusted mean) (95% CI)c | 0.0 (-0.9, 0.8) | 0.1 (-0.8, 0.9) | -- | -- | -- |
| Comparison vs linagliptin 5 mg (adjusted mean) (95% CI)c | -2.4 (-3.3, -1.5)d | -2.7 (-3.6, -1.8)d | -- | -- | -- |
a Full analysis population (observed case) using MMRM. MMRM model included treatment, renal function, region, visit, visit by treatment interaction, and baseline HbA1c.
b Patients with HbA1c above 7% at baseline: empagliflozin 25 mg/linagliptin 5 mg, n=123; empagliflozin 10 mg/linagliptin 5 mg, n=128; empagliflozin 25 mg, n=132; empagliflozin 10 mg, n=125; linagliptin 5 mg, n=119. Non-completers were considered failures (NCF).
c Full analysis population using last observation carried forward. ANCOVA model included treatment, renal function, region, baseline weight, and baseline HbA1c.
d p<0.001 for FPG; p<0.0001 for HbA1c and body weight
Figure 3. Adjusted Mean HbA1c Change at Each Time Point (Completers) and at Week 24 (mITT population):
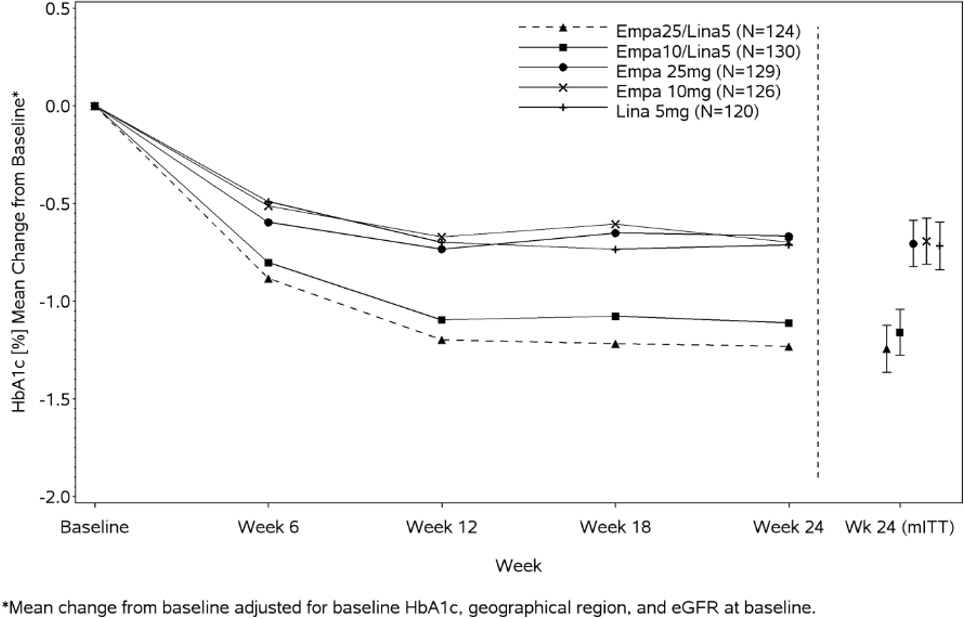
Empagliflozin Cardiovascular Outcome Study in Patients with Type 2 Diabetes Mellitus and Atherosclerotic Cardiovascular Disease
Empagliflozin is indicated to reduce the risk of cardiovascular death in adults with type 2 diabetes mellitus and established cardiovascular disease. The effect of empagliflozin on cardiovascular risk in adult patients with type 2 diabetes and established, stable, atherosclerotic cardiovascular disease is presented below.
The EMPA-REG OUTCOME study, a multicenter, multinational, randomized, double-blind parallel group trial compared the risk of experiencing a major adverse cardiovascular event (MACE) between empagliflozin and placebo when these were added to and used concomitantly with standard of care treatments for diabetes and atherosclerotic cardiovascular disease. Coadministered antidiabetic medications were to be kept stable for the first 12 weeks of the trial. Thereafter, antidiabetic and atherosclerotic therapies could be adjusted, at the discretion of investigators, to ensure participants were treated according to the standard care for these diseases.
A total of 7020 patients were treated (empagliflozin 10 mg = 2345; empagliflozin 25 mg = 2342; placebo = 2333) and followed for a median of 3.1 years. Approximately 72% of the study population was Caucasian, 22% was Asian, and 5% was Black. The mean age was 63 years and approximately 72% were male.
All patients in the study had inadequately controlled type 2 diabetes mellitus at baseline (HbA1c greater than or equal to 7%). The mean HbA1c at baseline was 8.1% and 57% of participants had diabetes for more than 10 years. Approximately 31%, 22% and 20% reported a past history of neuropathy, retinopathy and nephropathy to investigators, respectively and the mean eGFR was 74 mL/min/1.73 m². At baseline, patients were treated with one (~30%) or more (~70%) antidiabetic medications including metformin (74%), insulin (48%), sulfonylurea (43%) and dipeptidyl peptidase-4 inhibitor (11%).
All patients had established atherosclerotic cardiovascular disease at baseline including one (82%) or more (18%) of the following: a documented history of coronary artery disease (76%), stroke (23%) or peripheral artery disease (21%). At baseline, the mean systolic blood pressure was 136 mmHg, the mean diastolic blood pressure was 76 mmHg, the mean LDL was 86 mg/dL, the mean HDL was 44 mg/dL, and the mean urinary albumin to creatinine ratio (UACR) was 175 mg/g. At baseline, approximately 81% of patients were treated with renin angiotensin system inhibitors, 65% with beta-blockers, 43% with diuretics, 77% with statins, and 86% with antiplatelet agents (mostly aspirin).
The primary endpoint in EMPA-REG OUTCOME was the time to first occurrence of a Major Adverse Cardiac Event (MACE). A major adverse cardiac event was defined as occurrence of either a cardiovascular death or a non-fatal myocardial infarction (MI) or a non-fatal stroke. The statistical analysis plan had pre-specified that the 10 and 25 mg doses would be combined. A Cox proportional hazards model was used to test for non-inferiority against the pre-specified risk margin of 1.3 for the hazard ratio of MACE and superiority on MACE if non-inferiority was demonstrated. Type-1 error was controlled across multiples tests using a hierarchical testing strategy.
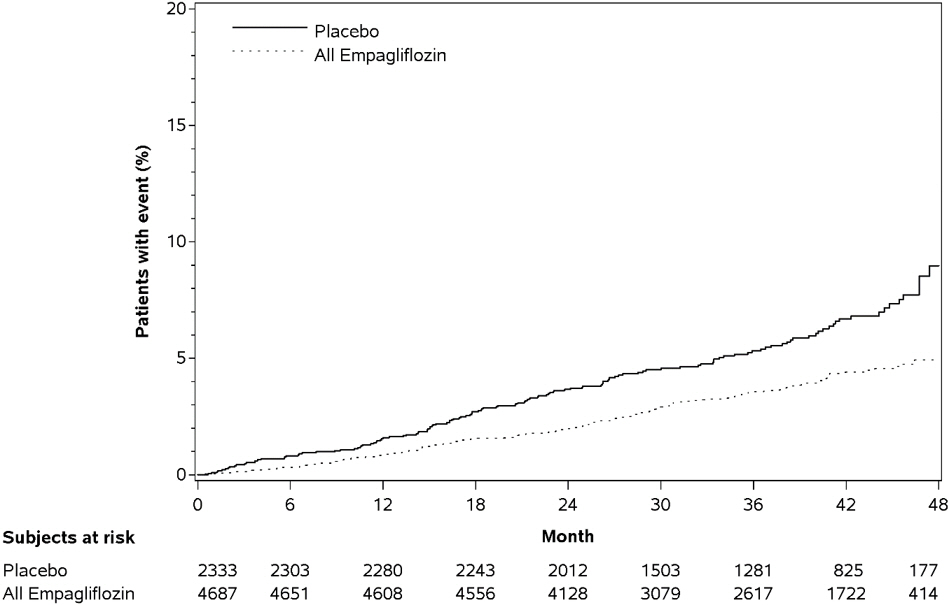
Empagliflozin significantly reduced the risk of first occurrence of primary composite endpoint of cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke (HR: 0.86; 95% CI: 0.74, 0.99). The treatment effect was due to a significant reduction in the risk of cardiovascular death in subjects randomized to empagliflozin (HR: 0.62; 95% CI: 0.49, 0.77), with no change in the risk of non-fatal myocardial infarction or non-fatal stroke (see Table 8 and Figures 4 and 5). Results for the 10 mg and 25 mg empagliflozin doses were consistent with results for the combined dose groups.
Table 8. Treatment Effect for the Primary Composite Endpoint and its Componentsa:
| Placebo N=2333 | Empagliflozin N=4687 | Hazard ratio vs placebo (95% CI) | |
|---|---|---|---|
| Composite of cardiovascular death, non-fatal myocardial infarction, non-fatal stroke (time to first occurrence)b | 282 (12.1%) | 490 (10.5%) | 0.86 (0.74, 0.99) |
| Non-fatal myocardial infarctionc | 121 (5.2%) | 213 (4.5%) | 0.87 (0.70, 1.09) |
| Non-fatal strokec | 60 (2.6%) | 150 (3.2%) | 1.24 (0.92, 1.67) |
| Cardiovascular deathc | 137 (5.9%) | 172 (3.7%) | 0.62 (0.49, 0.77) |
a Treated set (patients who had received at least one dose of study drug)
b p–value for superiority (2–sided) 0.04
c Total number of events
Figure 4. Estimated Cumulative Incidence of First MACE:
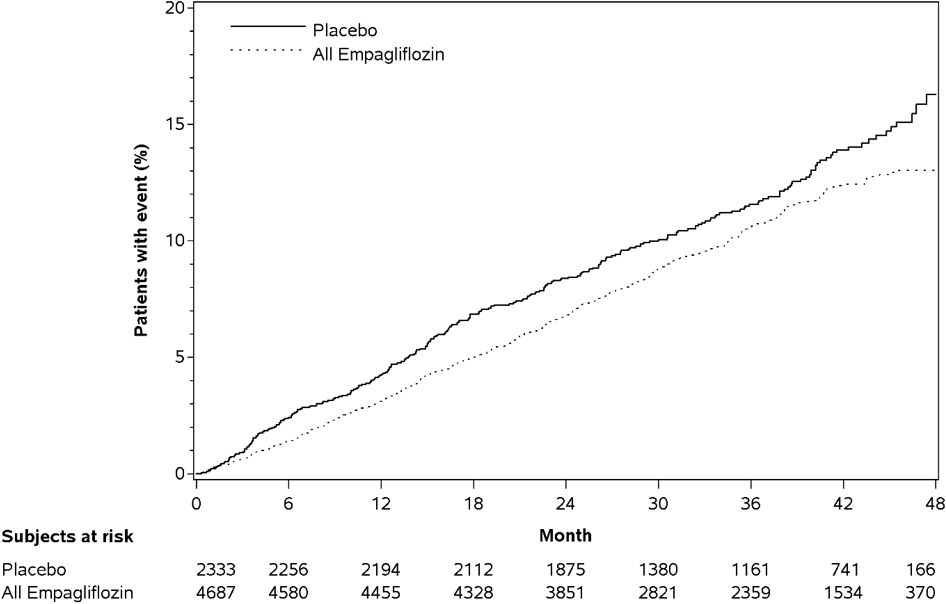
Figure 5. Estimated Cumulative Incidence of Cardiovascular Death:
The efficacy of empagliflozin on cardiovascular death was generally consistent across major demographic and disease subgroups.
Vital status was obtained for 99.2% of subjects in the trial. A total of 463 deaths were recorded during the EMPA-REG OUTCOME trial. Most of these deaths were categorized as cardiovascular deaths. The non-cardiovascular deaths were only a small proportion of deaths and were balanced between the treatment groups (2.1% in patients treated with empagliflozin, and 2.4% of patients treated with placebo).
Linagliptin Cardiovascular Safety Trials
CARMELINA
The cardiovascular risk of linagliptin was evaluated in CARMELINA (NCT0189753), a multinational, multi-center, placebo-controlled, double-blind, parallel group trial comparing linagliptin (N=3494) to placebo (N=3485) in adult patients with type 2 diabetes mellitus and a history of established macrovascular and/or renal disease. The trial compared the risk of major adverse cardiovascular events (MACE) between linagliptin and placebo when these were added to standard of care treatments for diabetes and other cardiovascular risk factors. The trial was event driven, the median duration of follow-up was 2.2 years and vital status was obtained for 99.7% of patients.
Patients were eligible to enter the trial if they were adults with type 2 diabetes, with HbA1c of 6.5% to 10%, and had either albuminuria and previous macrovascular disease (39% of enrolled population), or evidence of impaired renal function by eGFR and Urinary Albumin Creatinine Ratio (UACR) criteria (42% of enrolled population), or both (18% of enrolled population).
At baseline the mean age was 66 years and the population was 63% male, 80% Caucasian, 9% Asian, and 6% Black. Mean HbA1c was 8.0% and mean duration of type 2 diabetes mellitus was 15 years. The trial population included 17% patients ≥75 years of age and 62% patients with renal impairment defined as eGFR <60 mL/min/1.73 m². The mean eGFR was 55 mL/min/1.73 m² and 27% of patients had mild renal impairment (eGFR 60 to 90 mL/min/1.73 m²), 47% of patients had moderate renal impairment (eGFR 30 to <60 mL/min/1.73 m²) and 15% of patients had severe renal impairment (eGFR <30 mL/min/1.73 m²). Patients were taking at least one antidiabetic drug (97%), and the most common were insulin and analogues (57%), metformin (54%) and sulfonylurea (32%). Patients were also taking antihypertensives (96%), lipid lowering drugs (76%) with 72% on statin, and aspirin (62%).
The primary endpoint, MACE, was the time to first occurrence of one of three composite outcomes which included cardiovascular death, non-fatal myocardial infarction or non-fatal stroke. The study was designed as a non-inferiority trial with a pre-specified risk margin of 1.3 for the hazard ratio of MACE. A total of 434 patients on linagliptin and 420 patients on placebo experienced MACE. The incidence rate of MACE in both treatment arms: 56.3 MACE per 1000 patient-years on placebo vs. 57.7 MACE per 1000 patient-years on linagliptin. The estimated hazard ratio for MACE associated with linagliptin relative to placebo was 1.02 with a 95% confidence interval of (0.89, 1.17). The upper bound of this confidence interval, 1.17, excluded the risk margin of 1.3.
CAROLINA
The cardiovascular risk of linagliptin was evaluated in CAROLINA, a multi-center, multinational, randomized, double-blind parallel group trial comparing linagliptin (N=3023) to glimepiride (N=3010) in adult patients with type 2 diabetes mellitus and a history of established cardiovascular disease and/or multiple cardiovascular risk factors. The trial compared the risk of major adverse cardiovascular events (MACE) between linagliptin and glimepiride when these were added to standard of care treatments for diabetes and other cardiovascular risk factors. The trial was event driven, the median duration of follow-up was 6.23 years and vital status was obtained for 99.3% of patients.
Patients were eligible to enter the trial if they were adults with type 2 diabetes with insufficient glycemic control (defined as HbA1c of 6.5% to 8.5% or 6.5% to 7.5% depending on treatment-naïve, on monotherapy or on combination therapy), and were defined to be at high cardiovascular risk with previous vascular disease, evidence of vascular related end-organ damage, age ≥70 years, and/or two cardiovascular risk factors (duration of diabetes >10 years, systolic blood pressure >140 mmHg, current smoker, LDL cholesterol ≥135 mg/dL).
At baseline the mean age was 64 years and the population was 60% male, 73% Caucasian, 18% Asian, and 5% Black. The mean HbA1c was 7.15% and mean duration of type 2 diabetes was 7.6 years. The trial population included 34% patients ≥70 years of age and 19% patients with renal impairment defined as eGFR <60 mL/min/1.73 m². The mean eGFR was 77 mL/min/1.73 m². Patients were taking at least one antidiabetic drug (91%) and the most common were metformin (83%) and sulfonylurea (28%). Patients were also taking antihypertensives (89%), lipid lowering drugs (70%) with 65% on statin, and aspirin (47%).
The primary endpoint, MACE, was the time to first occurrence of one of three composite outcomes which included cardiovascular death, non-fatal myocardial infarction or non-fatal stroke. The study was designed as a non-inferiority trial with a pre-specified risk margin of 1.3 for the hazard ratio of MACE. A total of 356 patients on linagliptin and 362 patients on glimepiride experienced MACE. The incidence rate of MACE in both treatment arms: 20.7 MACE per 1000 patient-years on linagliptin vs. 21.2 MACE per 1000 patient-years on glimepiride. The estimated hazard ratio for MACE associated with linagliptin relative to glimepiride was 0.98 with a 95% confidence interval of (0.84, 1.14). The upper bound of this confidence interval, 1.14, excluded the risk margin of 1.3.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.