TRISENOX Concentrate for solution for infusion Ref.[6218] Active ingredients: Arsenic trioxide
Source: European Medicines Agency (EU) Revision Year: 2018 Publisher: Teva B.V., Swensweg 5, 2031 GA, Haarlem, Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: Other antineoplastic agents
ATC code: L01XX27
Mechanism of action
The mechanism of action of TRISENOX is not completely understood. Arsenic trioxide causes morphological changes and deoxyribonucleic acid (DNA) fragmentation characteristic of apoptosis in NB4 human promyelocytic leukaemia cells in vitro. Arsenic trioxide also causes damage or degradation of the fusion protein promyelocytic leukaemia/retinoic acid receptor-alpha (PML/RAR alpha).
Clinical efficacy and safety
Newly diagnosed non high risk APL patients
TRISENOX has been investigated in 77 newly diagnosed patients with low to intermediate risk APL, in a controlled, randomized, non-inferiority Phase 3 clinical study comparing the efficacy and safety of TRISENOX combined with all-trans-retinoic acid (ATRA) with those of ATRA + chemotherapy (eg, idarubicin and mitoxantrone) (Study APL0406). Patients with newly diagnosed APL confirmed by the presence of t(15; 17) or PML-RARα by RT-PCR or micro speckled PML nuclear distribution in leukaemic cells were included. No data are available on patient with variant translocations like t(11;17) (PLZF/RARα). Patients with significant arrhythmias, EKG abnormalities (congenital long QT syndrome, history or presence of significant ventricular or atrial tachyarrhythmia, clinically significant resting bradycardia (<50 beats per minute), QTc >450 msec on screening EKG, right bundle branch block plus left anterior hemiblock, bifascicular block) or neuropathy were excluded from the study. Patients in the ATRA + TRISENOX treatment group received oral ATRA at 45 mg/m² daily and IV TRISENOX at 0.15 mg/kg daily until CR. During consolidation, ATRA was given at the same dose for periods of 2 weeks on and 2 weeks off for a total of 7 courses, and TRISENOX was given at the same dose 5 days per week, 4 weeks on and 4 weeks off, for a total of 4 courses. Patients in the ATRA + chemotherapy treatment group received IV idarubicin at 12 mg/m² on days 2, 4, 6, and 8 and oral ATRA at 45 mg/m² daily until CR. During consolidation, patients received idarubicin at 5 mg/m² on days 1 to 4 and ATRA at 45 mg/m² daily for 15 days, then IV mitoxantrone at 10 mg/m² on days 1 12 to 5 and ATRA again at 45 mg/m² daily for 15 days, and finally a single dose of idarubicin at 12 mg/m² and ATRA at 45 mg/m² daily for 15 days. Each course of consolidation was initiated at haematological recovery from the previous course defined as absolute neutrophil count >1.5 × 109/l and platelets >100 × 109/l. Patients in the ATRA + chemotherapy treatment group also received maintenance treatment for up to 2 years, consisting of oral 6-mercaptopurine at 50 mg/m² daily, intramuscular methotrexate at 15 mg/m² weekly, and ATRA at 45 mg/m² daily for 15 days every 3 months.
The key efficacy results are summarised in table 3 below:
Table 3:
| Endpoint | ATRA + TRISENOX (n=77)[%] | ATRA + Chemotherapy (n=79)[%] | Confidence interval (CI) | P-value |
|---|---|---|---|---|
| 2-Year event-free survival (EFS) | 97 | 86 | 95% CI for the difference, 2-22 percentage points | p<0.001 for non-inferiority. p=0.02 for superiority of ATRA + TRISENOX |
| Haematologic complete remission (HCR) | 100 | 95 | p=0.12 | |
| 2-Year overall survival (OS) | 99 | 91 | p=0.02 | |
| 2-Year disease-free survival (DFS) | 97 | 90 | p=0.11 | |
| 2-Year cumulative incidence of relapse (CIR) | 1 | 6 | p=0.24 |
APL = acute promyelocytic leukaemia; ATRA = all-trans-retinoic acid
Relapsed/refractory APL
TRISENOX has been investigated in 52 APL patients, previously treated with an anthracycline and a retinoid regimen, in two open-label, single-arm, non-comparative studies. One was a single investigator clinical study (n=12) and the other was a multicentre, 9-institution study (n=40). Patients in the first study received a median dose of 0.16 mg/kg/day of TRISENOX (range 0.06 to 0.20 mg/kg/day) and patients in the multicentre study received a fixed dose of 0.15 mg/kg/day. TRISENOX was administered intravenously over 1 to 2 hours until the bone marrow was free of leukaemic cells, up to a maximum of 60 days. Patients with complete remission received consolidation therapy with TRISENOX for 25 additional doses over a 5 week period. Consolidation therapy began 6 weeks (range, 3-8) after induction in the single institution study and 4 weeks (range, 3-6) in the multicentre study. Complete remission (CR) was defined as the absence of visible leukaemic cells in the bone marrow and peripheral recovery of platelets and white blood cells.
Patients in the single centre study had relapsed following 1-6 prior therapy regimens and 2 patients had relapsed following stem cell transplantation. Patients in the multicentre study had relapsed following 1-4 prior therapy regimens and 5 patients had relapsed following stem cell transplantation. The median age in the single centre study was 33 years (age range 9 to 75). The median age in the multicentre study was 40 years (age range 5 to 73).
The results are summarised in the table 4 below.
Table 4:
| Single centre trial N=12 | Multicentre trial N=40 | |
|---|---|---|
| TRISENOX dose, mg/kg/day (median, range) | 0.16 (0.06-0.20) | 0.15 |
| Complete remission | 11 (92%) | 34 (85%) |
| Time to bone marrow remission (median) | 32 days | 35 days |
| Τime to CR (median) | 54 days | 59 days |
| 18-Month survival | 67% | 66% |
The single institution study included 2 paediatric patients (<18 years old), both of whom achieved CR. The multicentre trial included 5 paediatric patients (< 18 years old), 3 of whom achieved CR. No children of less than 5 years of age were treated.
In a follow-up treatment after consolidation, 7 patients in the single institution study and 18 patients in the multicentre study received further maintenance therapy with TRISENOX. Three patients from the single institution study and 15 patients from the multicentre study had stem cell transplants after completing TRISENOX. The Kaplan-Meier median CR duration for the single institution study is 14 months and has not been reached for the multicentre study. At last follow-up, 6 of 12 patients in the single institution study were alive with a median follow-up time of 28 months (range 25 to 29). In the multicentre study 27 of 40 patients were alive with a median follow-up time of 16 months (range 9 to 25). Kaplan-Meier estimates of 18-month survival for each study are shown below.
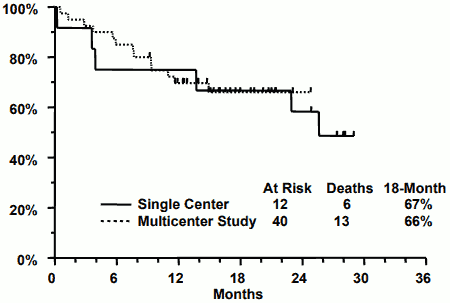
Cytogenetic confirmation of conversion to a normal genotype and reverse transcriptase - polymerase chain reaction (RT-PCR) detection of PML/RAR conversion to normal are shown in table 5 below.
Cytogenetics after TRISENOX therapy
Table 5:
| Single centre pilot trial N with CR=11 | Multicentre trial N with CR=34 | |
|---|---|---|
| Conventional Cytogenetics [t(15;17)] | ||
| Absent | 8 (73%) | 31 (91%) |
| Present | 1 (9%) | 0% |
| Not evaluable | 2 (18%) | 3 (9%) |
| RT-PCR for PML/RARα | ||
| Negative | 8 (73%) | 27 (79%) |
| Positive | 3 (27%) | 4 (12%) |
| Not evaluable | 0 | 3 (9%) |
Responses were seen across all age groups tested, ranging from 6 to 75 years. The response rate was similar for both genders. There is no experience on the effect of TRISENOX on the variant APL containing the t(11;17) and t(5;17) chromosomal translocations.
Paediatric population
The experience in children is limited. Of 7 patients under 18 years of age (range 5 to 16 years) treated with TRISENOX at the recommended dose of 0.15 mg/kg/day, 5 patients achieved a complete response (see section 4.2).
Pharmacokinetic properties
The inorganic, lyophilized form of arsenic trioxide, when placed into solution, immediately forms the hydrolysis product arsenious acid (AsIII). AsIII is the pharmacologically active species of arsenic trioxide.
Distribution
The volume of distribution (Vd for AsIII is large (>400 l) indicating significant distribution into the tissues with negligible protein binding. Vd is also weight dependent, increasing with increasing body weight. Total arsenic accumulates mainly in the liver, kidney, and heart and, to a lesser extent, in the lung, hair, and nails.
Biotransformation
The metabolism of arsenic trioxide involves oxidation of arsenious acid (AsIII), the active species of arsenic trioxide, to arsenic acid (AsV), as well as oxidative methylation to monomethylarsonic acid (MMAV) and dimethylarsinic acid (DMAV) by methyltransferases, primarily in the liver. The pentavalent metabolites, MMAV and DMAV, are slow to appear in plasma (approximately 10-24 hours after first administration of arsenic trioxide), but due to their longer half-life, accumulate more upon multiple dosing than does AsIII. The extent of accumulation of these metabolites is dependent on the dosing regimen. Approximate accumulation ranged from 1.4- to 8-fold following multiple as compared to single dose administration. AsV is present in plasma only at relatively low levels.
In vitro enzymatic studies with human liver microsomes revealed that arsenic trioxide has no inhibitory activity on substrates of the major cytochrome P450 enzymes such as 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5, 4A9/11. Substances that are substrates for these P450 enzymes are not expected to interact with TRISENOX.
Elimination
Approximately 15% of the administered TRISENOX dose is excreted in the urine as unchanged AsIII. The methylated metabolites of AsIII (MMAV, DMAV) are primarily excreted in the urine. The plasma concentration of AsIII declines from peak plasma concentration in a biphasic manner with a mean terminal elimination half-life of 10 to 14 hours. The total clearance of AsIII over the single-dose range of 7-32 mg (administered as 0.15 mg/kg) is 49 l/h and the renal clearance is 9 l/h. Clearance is not dependent on the weight of the subject or the dose administered over the dose range studied. The mean estimated terminal elimination half-lives of the metabolites MMAV and DMAV are 32 hours and 70 hours, respectively.
Renal impairment
Plasma clearance of AsIII was not altered in patients with mild renal impairment (creatinine clearance of 50-80 ml/min) or moderate renal impairment (creatinine clearance of 30-49 ml/min). The plasma clearance of AsIII in patients with severe renal impairment (creatinine clearance less than 30 ml/min) was 40% lower when compared with patients with normal renal function (see section 4.4).
Systemic exposure to MMAV and DMAV tended to be larger in patients with renal impairment; the clinical consequence of this is unknown but no increased toxicity was noted.
Hepatic impairment
Pharmacokinetic data from patients with hepatocellular carcinoma having mild to moderate hepatic impairment indicate that AsIII or AsV do not accumulate following twice-weekly infusions. No clear trend toward an increase in systemic exposure to AsIII, AsV, MMAV or DMAV was observed with decreasing level of hepatic function as assessed by dose-normalized (per mg dose) AUC.
Linearity / non-linearity
In the total single dose range of 7 to 32 mg (administered as 0.15 mg/kg), systemic exposure (AUC) appears to be linear. The decline from peak plasma concentration of AsIII occurs in a biphasic manner and is characterized by an initial rapid distribution phase followed by a slower terminal elimination phase. After administration at 0.15 mg/kg on a daily (n=6) or twice-weekly (n=3) regimen, an approximate 2-fold accumulation of AsIII was observed as compared to a single infusion. This accumulation was slightly more than expected based on single-dose results.
Preclinical safety data
Limited reproductive toxicity studies of arsenic trioxide in animals indicate embryotoxicity and teratogenicity (neural tube defects, anophthalmia and microphthalmia) at administration of 1-10 times the recommended clinical dose (mg/m²). Fertility studies have not been conducted with TRISENOX. Arsenic compounds induce chromosomal aberrations and morphological transformations of mammalian cells in vitro and in vivo. No formal carcinogenicity studies of arsenic trioxide have been performed. However, arsenic trioxide and other inorganic arsenic compounds are recognised as human carcinogens.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.