TRIZIVIR Film-coated tablet Ref.[107709] Active ingredients: Abacavir Lamivudine Zidovudine Zidovudine, Lamivudine and Abacavir
Source: European Medicines Agency (EU) Revision Year: 2022 Publisher: ViiV Healthcare BV, Van Asch van Wijckstraat 55H, 3811 LP Amersfoort, Netherlands
4.3. Contraindications
Hypersensitivity to the active substances or to any of the excipients listed in section 6.1. See sections 4.4 and 4.8.
Patients with end-stage renal disease.
Due to the active substance zidovudine, Trizivir is contraindicated in patients with abnormally low neutrophil counts (<0.75 x 109/l), or abnormally low haemoglobin levels (<7.5 g/dl or 4.65 mmol/l) (see section 4.4).
4.4. Special warnings and precautions for use
The special warnings and precautions relevant to abacavir, lamivudine and zidovudine are included in this section. There are no additional precautions or warnings relevant to the combination Trizivir.
Hypersensitivity Reactions (see also section 4.8 ) Abacavir is associated with a risk for hypersensitivity reactions (HSR) (see section4.8) characterised by fever and/or rash with other symptoms indicating multi-organ involvement. HSRs have been observed with abacavir, some of which have been life-threatening, and in rare cases fatal, when not managed appropriately.
The risk for abacavir HSR to occur is high for patients who test positive for the HLA-B*5701 allele. However, abacavir HSRs have been reported at a lower frequency in patients who do not carry this allele.
Therefore the following should be adhered to:
- HLA-B*5701 status must always be documented prior to initiating therapy.
- Trizivir should never be initiated in patients with a positive HLA-B*5701 status, nor in patients with a negative HLA-B*5701 status who had a suspected abacavir HSR on a previous abacavir-containing regimen. (e.g. Kivexa, Ziagen, Triumeq)
- Trizivir must be stopped without delay, even in the absence of the HLA-B*5701 allele, if an HSR is suspected. Delay in stopping treatment with Trizivir after the onset of hypersensitivity may result in a life-threatening reaction.
- After stopping treatment with Trizivir for reasons of a suspected HSR, Trizivir or any other medicinal product containing abacavir (e.g. Kivexa, Ziagen, Triumeq) must never be reinitiated.
- Restarting abacavir containing products following a suspected abacavir HSR can result in a prompt return of symptoms within hours. This recurrence is usually more severe than on initial presentation, and may include life-threatening hypotension and death.
- In order to avoid restarting abacavir patients who have experienced a suspected HSR should be instructed to dispose of their remaining Trizivir tablets
Clinical description of abacavir HSR
Abacavir HSR has been well characterised through clinical studies and during post marketing followup. Symptoms usually appeared within the first six weeks (median time to onset 11 days) of initiation of treatment with abacavir, although these reactions may occur at any time during therapy.
Almost all HSR to abacavir include fever and/or rash. Other signs and symptoms that have been observed as part of abacavir HSR are described in detail in section 4.8 (Description of selected adverse reactions), including respiratory and gastrointestinal symptoms. Importantly, such symptoms may lead to misdiagnosis of HSR as respiratory disease (pneumonia, bronchitis, pharyngitis), or gastroenteritis.
The symptoms related to HSR worsen with continued therapy and can be life-threatening. These symptoms usually resolve upon discontinuation of abacavir.
Rarely, patients who have stopped abacavir for reasons other than symptoms of HSR have also experienced life-threatening reactions within hours of re-initiating abacavir therapy (see Section 4.8 Description of selected adverse reactions). Restarting abacavir in such patients must be done in a setting where medical assistance is readily available.
Lactic acidosis
Lactic acidosis, usually associated with hepatomegaly and hepatic steatosis, has been reported with the use of zidovudine. Early symptoms (symptomatic hyperlactatemia) include benign digestive symptoms (nausea, vomiting and abdominal pain), non-specific malaise, loss of appetite, weight loss, respiratory symptoms (rapid and/or deep breathing), or neurological symptoms (including motor weakness).
Lactic acidosis has a high mortality and may be associated with pancreatitis, liver failure, or renal failure.
Lactic acidosis generally occurred after a few or several months of treatment.
Treatment with zidovudine should be discontinued in the setting of symptomatic hyperlactatemia and metabolic/lactic acidosis, progressive hepatomegaly, or rapidly elevating aminotransferase levels.
Caution should be exercised when administering zidovudine to any patient (particularly obese women) with hepatomegaly, hepatitis or other known risk factors for liver disease and hepatic steatosis (including certain medicinal products and alcohol). Patients co-infected with hepatitis C and treated with alpha interferon and ribavirin may constitute a special risk.
Patients at increased risk should be followed closely.
Mitochondrial dysfunction following exposure in utero
Nucleoside and nucleotide analogues may impact mitochondrial function to a variable degree, which is most pronounced with stavudine, didanosine and zidovudine. There have been reports of mitochondrial dysfunction in HIV-negative infants exposed in utero and/or post-natally to nucleoside analogues; these have predominantly concerned treatment with regimens containing zidovudine. The main adverse reactions reported are haematological disorders (anaemia, neutropenia), and metabolic disorders (hyperlactatemia, hyperlipasemia). These adverse reactions have often been transitory. Late onset neurological disorders have been reported rarely (hypertonia, convulsion, abnormal behaviour). Whether such neurological disorders are transient or permanent is currently unknown. These findings should be considered for any child exposed in utero to nucleotide and nucleotide analogues, who presents with severe clinical findings of unknown etiology, particularly neurologic findings. These findings do not affect current national recommendations to use antiretroviral therapy in pregnant women to prevent vertical transmission of HIV.
Lipoatrophy
Treatment with zidovudine has been associated with loss of subcutaneous fat, which has been linked to mitochondrial toxicity. The incidence and severity of lipoatrophy are related to cumulative exposure. This fat loss, which is most evident in the face, limbs and buttocks, may not be reversible when switching to a zidovudine-free regimen. Patients should be regularly assessed for signs of lipoatrophy during therapy with zidovudine and zidovudine-containing products (Combivir and Trizivir). Therapy should be switched to an alternative regimen if there is suspicion of lipoatrophy development.
Weight and metabolic parameters
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Haematological adverse reactions
Anaemia, neutropenia and leukopenia (usually secondary to neutropenia) can be expected to occur in patients receiving zidovudine. These occurred more frequently at higher zidovudine doses (1200- 1500 mg/day) and in patients with poor bone marrow reserve prior to treatment, particularly with advanced HIV disease. Haematological parameters should therefore be carefully monitored (see section 4.3) in patients receiving Trizivir. These haematological effects are not usually observed before four to six week's therapy. For patients with advanced symptomatic HIV disease, it is generally recommended that blood tests are performed at least every two weeks for the first three months of therapy and at least monthly thereafter.
In patients with early HIV disease haematological adverse reactions are infrequent. Depending on the overall condition of the patient, blood tests may be performed less often, for example every one to three months. Additionally, dose adjustment of zidovudine may be required if severe anaemia or myelosuppression occurs during treatment with Trizivir, or in patients with pre-existing bone marrow compromise e.g. haemoglobin <9 g/dl (5.59 mmol/l) or neutrophil count <1.0 x 109/l (see section 4.2). As dose adjustment of Trizivir is not possible separate preparations of zidovudine, abacavir and lamivudine should be used. Physicians should refer to the individual prescribing information for these medicinal products.
Pancreatitis
Cases of pancreatitis have occurred rarely in patients treated with abacavir, lamivudine and zidovudine. However, it is not clear whether these cases were due to treatment with these medicinal products or to the underlying HIV disease. Treatment with Trizivirshould be stopped immediately if clinical signs, symptoms or laboratory abnormalities suggestive of pancreatitis occur.
Liver disease
If lamivudine is being used concomitantly for the treatment of HIV and hepatitis B virus (HBV) infection, additional information relating to the use of lamivudine in the treatment of HBV is available in the Zeffix SmPC.
The safety and efficacy of Trizivir has not been established in patients with significant underlying liver disorders. Trizivir is not recommended in patients with moderate or severe hepatic impairment (see sections 4.2 and 5.2).
Patients with chronic hepatitis B or C and treated with combination antiretroviral therapy are at an increased risk of severe and potentially fatal hepatic adverse reactions. In case of concomitant antiviral therapy for hepatitis B or C, please refer also to the relevant product information for these medicinal products.
If Trizivir is discontinued in patients co-infected with hepatitis B virus, periodic monitoring of both liver function tests and markers of HBV replication is recommended, as withdrawal of lamivudine may result in an acute exacerbation of hepatitis (see Zeffix SmPC).
Patients with pre-existing liver dysfunction including chronic active hepatitis have an increased frequency of liver function abnormalities during combination antiretroviral therapy and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered.
Patients co-infected with hepatitis B or C virus
The concomitant use of ribavirin with zidovudine is not recommended due to an increased risk of anaemia (see section 4.5).
Children and adolescents
Because insufficient data are available, the use of Trizivir in children or adolescents is not recommended. In this patient population, hypersensitivity reactions are particularly difficult to identify.
Immune Reactivation Syndrome
In HIV-infected patients with severe immune deficiency at the time of institution of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterium infections, and Pneumocystis jirovecii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and can occur many months after initiation of treatment.
Osteonecrosis
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported particularly in patients with advanced HIV-disease and/or long-term exposure to combination antiretroviral therapy (CART). Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Opportunistic infections
Patients should be advised that Trizivir or any other antiretroviral therapy does not cure HIV infection and that they may still develop opportunistic infections and other complications of HIV infection. Therefore, patients should remain under close clinical observation by physicians experienced in the treatment of these associated HIV diseases.
Cardiovascular events
Although the available data from clinical and observational studies with abacavir show inconsistent results, several studies suggest an increased risk of cardiovascular events (notably myocardial infarction) in patients treated with abacavir. Therefore, when prescribing Trizivir, action should be taken to minimize all modifiable risk factors (e.g. smoking, hypertension, and hyperlipidaemia). In addition, alternative treatment options to the abacavir containing regimen should be considered when treating patients with a high cardiovascular risk.
Administration in subjects with moderate renal impairment
Patients with a creatinine clearance between 30 and 49 mL/min receiving Trizivir may experience a 1.6-to 3.3-fold higher lamivudine exposure (AUC) than patients with a creatinine clearance ≥50 mL/min. There are no safety data from randomized, controlled trials comparing Trizivir to the individual components in patients with a creatinine clearance between 30 and 49 mL/min who received dose-adjusted lamivudine. In the original lamivudine registrational trials in combination with zidovudine, higher lamivudine exposures were associated with higher rates of haematologic toxicities (neutropenia and anaemia), although discontinuations due to neutropenia or anaemia each occurred in <1% of subjects. Other lamivudine-related adverse events (such as gastro-intestinal and hepatic disorders) may occur.
Patients with a sustained creatinine clearance between 30 and 49 mL/min who receive Trizivir should be monitored for lamivudine-related adverse events, notably haematologic toxicities. If new or worsening neutropenia or anaemia develop, a dose adjustment of lamivudine, per lamivudine prescribing information, is indicated, which cannot be achieved with Trizivir. Trizivir should be discontinued and the individual components should be used to construct the treatment regimen.
Drug Interactions
To date there are insufficient data on the efficacy and safety of Trizivir given concomitantly with nonnucleoside reverse transcriptase inhibitors (NNRTIs) or the protease inhibitors (PIs) (see section 5.1).
Trizivir should not be taken with any other medicinal products containing lamivudine or medicinal products containing emtricitabine.
The concomitant use of stavudine with zidovudine should be avoided (see section 4.5).
The combination of lamivudine with cladribine is not-recommended (see section 4.5).
Excipients
This medicine contains less than 1 mmol sodium (23 mg) per dosage unit, that is to say essentially 'sodium-free'.
4.5. Interaction with other medicinal products and other forms of interaction
Trizivir contains abacavir, lamivudine and zidovudine, therefore any interactions identified for these individually are relevant to Trizivir. Clinical studies have shown that there are no clinically significant interactions between abacavir, lamivudine and zidovudine.
Abacavir is metabolised by UDP-glucuronyltransferase (UGT) enzymes and alcohol dehydrogenase; co-administration of inducers or inhibitors of UGT enzymes or with compounds eliminated through alcohol dehydrogenase could alter abacavir exposure. Zidovudine is primarily metabolised by UGT enzymes; co-administration of inducers or inhibitors of UGT enzymes could alter zidovudine exposure. Lamivudine is cleared renally. Active renal secretion of lamivudine in the urine is mediated through organic cation transporters (OCTs); co-administration of lamivudine with OCT inhibitors may increase lamivudine exposure.
Abacavir, lamivudine and zidovudine are not significantly metabolised by cytochrome P450 enzymes (such as CYP 3A4, CYP 2C9 or CYP 2D6) nor do they induce this enzyme system. Lamivudine and zidovudine do not inhibit cytochrome P450 enzymes. Abacavir shows limited potential to inhibit metabolism mediated by CYP3A4 and has been shown in vitro not to inhibit CYP2C9 or CYP 2D6 enzymes. In vitro studies have shown that abacavir has potential to inhibit cytochrome P450 1A1 (CYP1A1). Therefore, there is little potential for interactions with antiretroviral protease inhibitors, non-nucleosides and other medicinal products metabolised by major P450 enzymes.
Interaction studies have only been performed in adults. The list below should not be considered exhaustive but is representative of the classes studied.
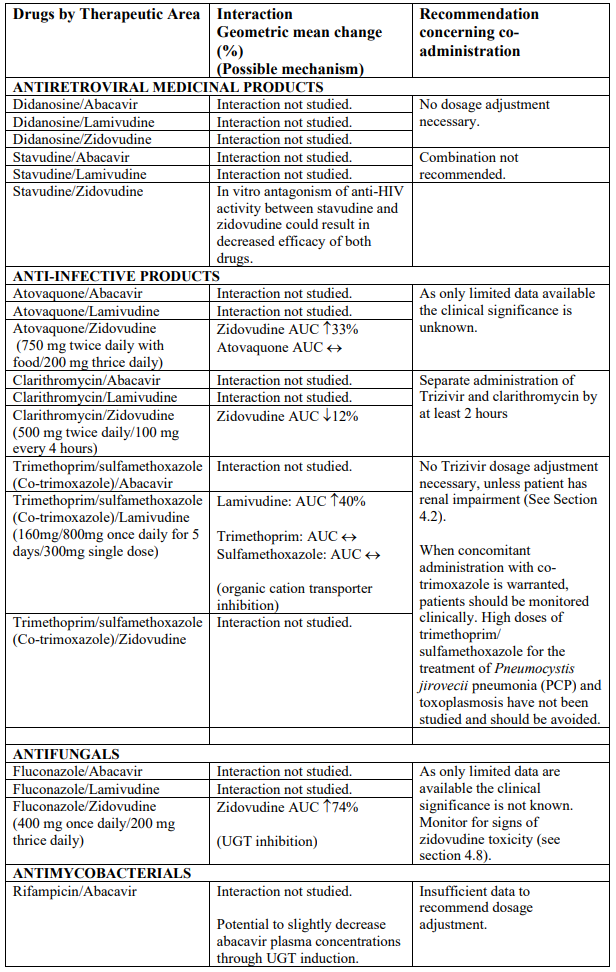
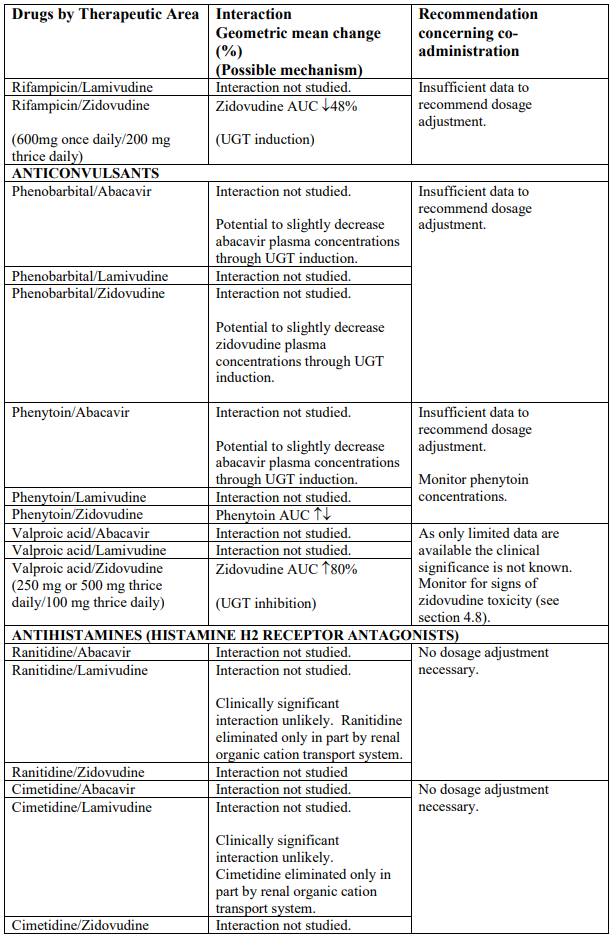
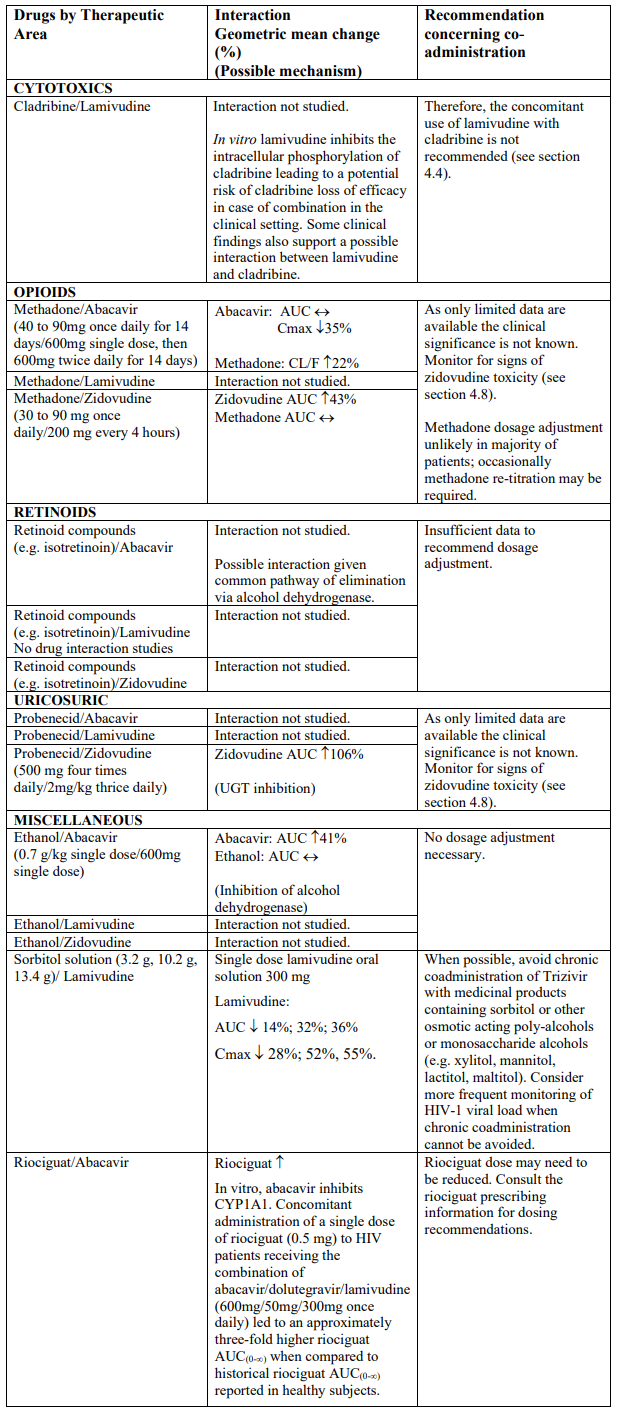
Abbreviations: ↑ = Increase; ↓ = decrease; ↔ = no significant change; AUC = area under the concentration versus time curve; Cmax = maximum observed concentration; CL/F = apparent oral clearance
Exacerbation of anaemia due to ribavirin has been reported when zidovudine is part of the regimen used to treat HIV although the exact mechanism remains to be elucidated. The concomitant use of ribavirin with zidovudine is not recommended due to an increased risk of anaemia (see section 4.4). Consideration should be given to replacing zidovudine in a combination ART regimen if this is already established. This would be particularly important in patients with a known history of zidovudine induced anaemia.
Concomitant treatment, especially acute therapy, with potentially nephrotoxic or myelosuppressive medicinal products (e.g. systemic pentamidine, dapsone, pyrimethamine, co-trimoxazole, amphotericin, flucytosine, ganciclovir, interferon, vincristine, vinblastine and doxorubicin) may also increase the risk of adverse reactions to zidovudine (see section 4.8). If concomitant therapy with Trizivir and any of these medicinal products is necessary then extra care should be taken in monitoring renal function and haematological parameters and, if required, the dosage of one or more agents should be reduced.
Limited data from clinical trials do not indicate a significantly increased risk of adverse reactions to zidovudine with cotrimoxazole (see interaction information above relating to lamivudine and cotrimoxazole), aerosolised pentamidine, pyrimethamine and acyclovir at doses used in prophylaxis.
4.6. Fertility, pregnancy and lactation
Pregnancy
As a general rule, when deciding to use antiretroviral agents for the treatment of HIV infection in pregnant women and consequently for reducing the risk of HIV vertical transmission to the newborn, he animal data as well as the clinical experience in pregnant women should be taken into account. In the present case, the use in pregnant women of zidovudine, with subsequent treatment of the newborn infants, has been shown to reduce the rate of maternal-foetal transmission of HIV. There are no data on the use of Trizivir in pregnancy. A moderate amount of data on pregnant women taking the individual actives abacavir, lamivudine and zidovudine in combination indicates no malformative toxicity (more than 300 outcomes from first trimester exposures). A large amount of data on pregnant women taking lamivudine or zidovudine indicate no malformative toxicity (more than 3000 outcomes from first trimester exposure each, of which over 2000 outcomes involved exposure to both lamivudine and zidovudine). Moderate amount of data (more than 600 outcomes from first trimester) indicates no malformative toxicity for abacavir. The malformative risk is unlikely in humans based on the mentioned moderate amount of data.
The active ingredients of Trizivir may inhibit cellular DNA replication, zidovudine has been shown to be transplacental carcinogen in one animal study and abacavir has been shown to be carcinogenic in animal models (see section 5.3). The clinical relevance of these findings is unknown.
For patients co-infected with hepatitis who are being treated with a lamivudine containing medicinal product such as Trizivir and subsequently become pregnant, consideration should be given to the possibility of a recurrence of hepatitis on discontinuation of lamivudine.
Mitochondrial dysfunction
Nucleoside and nucleotide analogues have been demonstrated in vitro and in vivo to cause a variable degree of mitochondrial damage. There have been reports of mitochondrial dysfunction in HIV-negative infants exposed in utero and/or post-natally to nucleoside analogues (see section 4.4).
Breast-feeding
Abacavir and its metabolites are excreted into the milk of lactating rats. Abacavir is also excreted into human milk.
Based on more than 200 mother/child pairs treated for HIV, serum concentrations of lamivudine in breastfed infants of mothers treated for HIV are very low (<4% of maternal serum concentrations) and progressively decrease to undetectable levels when breastfed infants reach 24 weeks of age. There are no data available on the safety of abacavir and lamivudine when administered to babies less than three months old.
After administration of a single dose of 200 mg zidovudine to HIV-infected women, the mean concentration of zidovudine was similar in human milk and serum.
It is recommended that women living with HIV do not breast-feed their infants in order to avoid transmission of HIV.
Fertility
Studies in animals showed that neither abacavir nor lamivudine nor zidovudine had any effect on fertility (see section 5.3). Zidovudine has been shown not to affect the number of sperm, sperm morphology and motility in man.
4.7. Effects on ability to drive and use machines
No studies on the effects on the ability to drive and use machines have been performed. The clinical status of the patient and the adverse event profile of Trizivir should be borne in mind when considering the patient's ability to drive or operate machinery.
4.8. Undesirable effects
Summary of the safety profile
Adverse reactions have been reported with abacavir, lamivudine and zidovudine used separately or in combination for therapy of HIV disease. Because Trizivir contains abacavir, lamivudine and zidovudine, the adverse reactions associated with these compounds may be expected.
Tabulated list of adverse reactions reported with the individual substances
The adverse reactions reported with abacavir, lamivudine and zidovudine are presented in Table 1. They are listed by body system, organ class and absolute frequency. Frequencies are defined as very common (>1/10), common (>1/100 to <1/10), uncommon (>1/1000 to <1/100), rare (>1/10,000 to <1/1000), very rare (<1/10,000). Care must be taken to eliminate the possibility of a hypersensitivity reaction if any of these symptoms occur.
Table 1. Adverse reactions reported with the individual components of Trizivir:
| Abacavir | Lamivudine | Zidovudine | |||
|---|---|---|---|---|---|
| IMPORTANT: for information on abacavir hypersensitivity see the information below, under the Description of selected adverse reactions Abacavir hypersensitivity | |||||
| Blood and lymphatic system disorders | |||||
| Uncommon: neutropenia, anaemia (both occasionally severe), thrombocytopenia Very rare: pure red cell aplasia | Common: anaemia, neutropenia and leukopenia Uncommon: thrombocytopenia and pancytopenia with marrow hypoplasia Rare: pure red cell aplasia Very rare: aplastic anaemia | ||||
| Immune system disorders | |||||
| Common: hypersensitivity | |||||
| Metabolism and nutrition disorders | |||||
| Common: anorexia Very Rare : lactic acidosis | Very Rare : lactic acidosis | Rare: anorexia, lactic acidosis in the absence of hypoxaemia | |||
| Psychiatric disorders | |||||
| Rare: anxiety, depression | Nervous system disorders | ||||
| Common: headache | Common: headache, insomnia Very rare: peripheral neuropathy (paraesthesiae) | Very common: headache Common: dizziness Rare: insomnia, paraesthesia, somnolence, loss of mental acuity, convulsions | |||
| Cardiac disorders | |||||
| Rare: cardiomyopathy | |||||
| Respiratory, thoracic and mediastinal disorders | |||||
| Common: cough, nasal symptoms | Uncommon: dyspnoea Rare: cough | ||||
| Gastrointestinal disorders | |||||
| Common: nausea, vomiting, diarrhoea Rare: pancreatitis | Common: nausea, vomiting, abdominal pain, diarrhoea Rare: rises in serum amylase, pancreatitis | Very common: Nausea Common: vomiting, abdominal pain, and diarrhoea Uncommon: flatulence Rare: oral mucosa pigmentation, taste disturbance dyspepsia, pancreatitis | |||
| Hepatobiliary disorders | |||||
| Uncommon: transient rises in liver enzymes (AST, ALT) Rare: hepatitis | Common: raised blood levels of liver enzymes and bilirubin Rare: liver disorders such as severe hepatomegaly with steatosis | ||||
| Skin and subcutaneous tissue disorders | |||||
| Common: rash (without systemic symptoms) Very rare: erythema multiforme, Stevens-Johnson syndrome and toxic epidermal necrolysis | Common: rash, alopecia | Uncommon: rash and pruritus Rare: nail and skin pigmentation, urticaria and sweating | |||
| Musculoskeletal and connective tissue disorders | |||||
| Common: arthralgia, muscle disorders Rare: rhabdomyolysis | Common: myalgia Uncommon: myopathy | ||||
| Renal and urinary disorders | |||||
| Rare: urinary frequency | |||||
| Reproductive system and breast disorders | |||||
| Rare: gynaecomastia | |||||
| General disorders and administration site conditions | |||||
| Common: fever, lethargy, fatigue | Common: fatigue, malaise, fever | Common: malaise Uncommon: fever, generalised pain and asthenia Rare: chills, chest pain, and influenza-like syndrome | |||
Many of the adverse reactions listed in the table occur commonly (nausea, vomiting, diarrhoea, fever, lethargy, rash) in patients with abacavir hypersensitivity. Therefore, patients with any of these symptoms should be carefully evaluated for the presence of this hypersensitivity (see section 4.4). Very rarely cases of erythema multiforme, Stevens-Johnson syndrome or toxic epidermal necrolysis have been reported where abacavir hypersensitivity could not be ruled out. In such cases medicinal products containing abacavir should be permanently discontinued.
Description of selected adverse reactions
Abacavir hypersensitivity
The signs and symptoms of this HSR are listed below. These have been identified either from clinical studies or post marketing surveillance. Those reported in at least 10% of patients with a hypersensitivity reaction are in bold text.
Almost all patients developing hypersensitivity reactions will have fever and/or rash (usually maculopapular or urticarial) as part of the syndrome, however reactions have occurred without rash or fever. Other key symptoms include gastrointestinal, respiratory or constitutional symptoms such as lethargy and malaise.
Skin: Rash (usually maculopapular or urticarial)
Gastrointestinal tract: Nausea, vomiting, diarrhoea, abdominal pain, mouth ulceration
Respiratory tract: Dyspnoea, cough, sore throat, adult respiratory distress syndrome, respiratory failure
Miscellaneous: Fever, lethargy, malaise, oedema, lymphadenopathy, hypotension, conjunctivitis, anaphylaxis
Neurological/Psychiatry: Headache, paraesthesia
Haematological: Lymphopenia
Liver/pancreas: Elevated liver function tests, hepatitis, hepatic failure
Musculoskeletal: Myalgia, rarely myolysis, arthralgia, elevated creatine phosphokinase
Urology: Elevated creatinine, renal failure
Symptoms related to this HSR worsen with continued therapy and can be life- threatening and in rare instance, have been fatal.
Restarting abacavir following an abacavir HSR results in a prompt return of symptoms within hours. This recurrence of the HSR is usually more severe than on initial presentation, and may include life-threatening hypotension and death. Similar reactions have also occurred infrequently after restarting abacavir in patients who had only one of the key symptoms of hypersensitivity (see above) prior to stopping abacavir; and on very rare occasions have also been seen in patients who have restarted therapy with no preceding symptoms of a HSR (i.e., patients previously considered to be abacavir tolerant).
Haematological adverse reactions with zidovudine
Anaemia, neutropenia and leukopenia occurred more frequently at higher doses (1,200-1,500 mg/day) and in patients with advanced HIV disease (especially when there is poor bone marrow reserve prior to treatment) and particularly in patients with CD4 cell counts less than 100/mm³. Dose reduction or cessation of therapy may become necessary (see section 4.4). The anaemia may necessitate transfusions.
The incidence of neutropenia was also increased in those patients whose neutrophil counts, haemoglobin levels and serum vitamin B12 levels were low at the start of zidovudine therapy.
Lactic acidosis
Treatment with zidovudine has been associated with cases of lactic acidosis, sometimes fatal, usually associated with severe hepatomegaly and hepatic steatosis, (see section 4.4).
Lipoatrophy
Treatment with zidovudine has been associated with loss of subcutaneous fat which is most evident in the face, limbs and buttocks. Patients receiving Trizivir should be frequently examined and questioned for signs of lipoatrophy. When such development is found, treatment with Trizivir should not be continued (see section 4.4).
Metabolic parameters
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
Immune Reactivation Syndrome
In HIV-infected patients with severe immune deficiency at the time of initiation of combination antiretroviral therapy (CART), an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Osteonecrosis
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to combination antiretroviral therapy (CART). The frequency of this is unknown (see section 4.4).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
6.2. Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.