ULTRAM Coated tablet Ref.[10616] Active ingredients: Tramadol
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
ULTRAM contains tramadol, an opioid agonist and inhibitor of norepinephrine and serotonin re-uptake. Although the mode of action is not completely understood, the analgesic effect of tramadol is believed to be due to both binding to µ-opioid receptors and weak inhibition of re-uptake of norepinephrine and serotonin.
Opioid activity is due to both low affinity binding of the parent compound and higher affinity binding of the O-demethylated metabolite M1 to µ-opioid receptors. In animal models, M1 is up to 6 times more potent than tramadol in producing analgesia and 200 times more potent in µ-opioid binding. Tramadol-induced analgesia is only partially antagonized by the opioid antagonist naloxone in several animal tests. The relative contribution of both tramadol and M1 to human analgesia is dependent upon the plasma concentrations of each compound [see Clinical Pharmacology (12.2)].
Analgesia in humans begins approximately within one hour after administration and reaches a peak in approximately two to three hours.
12.2. Pharmacodynamics
Effects on the Central Nervous System
Tramadol produces respiratory depression by direct action on brain stem respiratory centers. The respiratory depression involves a reduction in the responsiveness of the brain stem respiratory centers to both increases in carbon dioxide tension and electrical stimulation.
Tramadol administration may produce a constellation of symptoms including nausea and vomiting, dizziness, and somnolence.
Tramadol causes miosis, even in total darkness. Pinpoint pupils are a sign of opioid overdose but are not pathognomonic (e.g., pontine lesions of hemorrhagic or ischemic origins may produce similar findings). Marked mydriasis rather than miosis may be seen due to hypoxia in overdose situations.
Effects on the Gastrointestinal Tract and Other Smooth Muscle
Tramadol causes a reduction in motility associated with an increase in smooth muscle tone in the antrum of the stomach and duodenum. Digestion of food in the small intestine is delayed and propulsive contractions are decreased. Propulsive peristaltic waves in the colon are decreased, while tone may be increased to the point of spasm resulting in constipation. Other opioid-induced effects may include a reduction in biliary and pancreatic secretions, spasm of sphincter of Oddi, and transient elevations in serum amylase.
Effects on the Cardiovascular System
Tramadol produces peripheral vasodilation, which may result in orthostatic hypotension or syncope. Manifestations of peripheral vasodilation may include pruritus, flushing, red eyes, sweating and/or orthostatic hypotension.
The effect of oral tramadol on the QTcF interval was evaluated in a double-blind, randomized, four-way crossover, placebo- and positive- (moxifloxacin) controlled study in 68 adult male and female healthy subjects. At a 600 mg/day dose (1.5-fold the maximum immediate-release daily dose), the study demonstrated no significant effect on the QTcF interval.
Effects on the Endocrine System
Opioids inhibit the secretion of adrenocorticotropic hormone (ACTH), cortisol, and luteinizing hormone (LH) in humans. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon [see Warnings and Precautions (5.11); Adverse Reactions (6)].
Chronic use of opioids may influence the hypothalamic-pituitary-gonadal axis, leading to androgen deficiency that may manifest as low libido, impotence, erectile dysfunction, amenorrhea, or infertility. The causal role of opioids in the clinical syndrome of hypogonadism is unknown because the various medical, physical, lifestyle, and psychological stressors that may influence gonadal hormone levels have not been adequately controlled for in studies conducted to date [see Adverse Reactions (6)].
Effects on the Immune System
Opioids have been shown to have a variety of effects on components of the immune system in in vitro and animal models. The clinical significance of these findings is unknown. Overall, the effects of opioids appear to be modestly immunosuppressive.
Concentration–Efficacy Relationships
The minimum effective analgesic concentration will vary widely among patients, especially among patients who have been previously treated with potent opioid agonists. The minimum effective analgesic concentration of tramadol for any individual patient may increase over time due to an increase in pain, the development of a new pain syndrome and/or the development of analgesic tolerance [see Dosage and Administration (2)].
Concentration–Adverse Reaction Relationships
There is a relationship between increasing tramadol plasma concentration and increasing frequency of dose-related opioid adverse reactions such as nausea, vomiting, CNS effects, and respiratory depression. In opioid-tolerant patients, the situation may be altered by the development of tolerance to opioid-related adverse reactions [see Dosage and Administration (2)].
12.3. Pharmacokinetics
The analgesic activity of ULTRAM is due to both parent drug and the M1 metabolite [see Clinical Pharmacology (12.1,12.2)]. Tramadol is administered as a racemate and both the [-] and [+] forms of both tramadol and M1 are detected in the circulation. Linear pharmacokinetics have been observed following multiple doses of 50 and 100 mg to steady-state.
Absorption
The mean absolute bioavailability of a 100 mg oral dose is approximately 75%. The mean peak plasma concentration of racemic tramadol and M1 occurs at two and three hours, respectively, after administration in healthy adults. In general, both enantiomers of tramadol and M1 follow a parallel time course in the body following single and multiple doses although small differences (~ 10%) exist in the absolute amount of each enantiomer present.
Steady-state plasma concentrations of both tramadol and M1 are achieved within two days with four times per day dosing. There is no evidence of self-induction (see Figure 1 and Table 3 below).
Figure 1. Mean Tramadol and M1 Plasma Concentration Profiles after a Single 100 mg Oral Dose and after Twenty-Nine 100 mg Oral Doses of Tramadol HCl given four times per day:
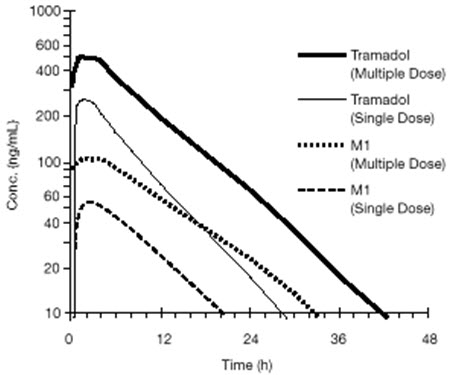
Table 3. Mean (%CV) Pharmacokinetic Parameters for Racemic Tramadol and M1 Metabolite:
| Population/Dosage Regimen* | Parent Drug/Metabolite | Peak Conc. (ng/mL) | Time to Peak (hrs) | Clearance/F† (mL/min/Kg) | t1/2 (hrs) |
|---|---|---|---|---|---|
| Healthy Adults, 100 mg qid, MD p.o. | Tramadol M1 | 592 (30) 110 (29) | 2.3 (61) 2.4 (46) | 5.90 (25)‡ | 6.7 (15) 7.0 (14) |
| Healthy Adults, 100 mg SD p.o. | Tramadol M1 | 308 (25) 55.0 (36) | 1.6 (63) 3.0 (51) | 8.50 (31)‡ | 5.6 (20) 6.7 (16) |
| Geriatric, (>75 yrs) 50 mg SD p.o. | Tramadol M1 | 208 (31)§ | 2.1 (19)§ | 6.89 (25)‡ | 7.0 (23)§ |
| Hepatic Impaired, 50 mg SD p.o. | Tramadol M1 | 217 (11) 19.4 (12) | 1.9 (16) 9.8 (20) | 4.23 (56)‡ | 13.3 (11) 18.5 (15) |
| Renal Impaired, CLcr10–30 mL/min 100 mg SD i.v. | Tramadol M1 | ‡ ‡ | ‡ ‡ | 4.23 (54)‡ | 10.6 (31) 11.5 (40) |
| Renal Impaired, CLcr<5 mL/min 100 mg SD i.v. | Tramadol M1 | ‡ ‡ | ‡ ‡ | 3.73 (17)‡ | 11.0 (29) 16.9 (18) |
* SD = Single dose, MD = Multiple dose, p.o.= Oral administration, i.v.= Intravenous administration, q.i.d. = Four times daily
† F represents the oral bioavailability of tramadol
‡ Not applicable
§ Not measured
Food Effects
Oral administration of ULTRAM with food does not significantly affect its rate or extent of absorption, therefore, ULTRAM can be administered without regard to food.
Distribution
The volume of distribution of tramadol was 2.6 and 2.9 liters/kg in male and female subjects, respectively, following a 100 mg intravenous dose. The binding of tramadol to human plasma proteins is approximately 20% and binding also appears to be independent of concentration up to 10 mcg/mL. Saturation of plasma protein binding occurs only at concentrations outside the clinically relevant range.
Elimination
Tramadol is eliminated primarily through metabolism by the liver and the metabolites are eliminated primarily by the kidneys. The mean (%CV) apparent total clearance of tramadol after a single 100 mg oral dose is 8.50 (31) mL/min/kg. The mean terminal plasma elimination half-lives of racemic tramadol and racemic M1 are 6.3 ± 1.4 and 7.4 ± 1.4 hours, respectively. The plasma elimination half-life of racemic tramadol increased from approximately six hours to seven hours upon multiple dosing.
Metabolism
Tramadol is extensively metabolized after oral administration by a number of pathways, including CYP2D6 and CYP3A4, as well as by conjugation of parent and metabolites. Approximately 30% of the dose is excreted in the urine as unchanged drug, whereas 60% of the dose is excreted as metabolites. The remainder is excreted either as unidentified or as unextractable metabolites. The major metabolic pathways appear to be N- and O-demethylation and glucuronidation or sulfation in the liver. One metabolite (O-desmethyltramadol, denoted M1) is pharmacologically active in animal models. Formation of M1 is dependent on CYP2D6 and as such is subject to inhibition, which may affect the therapeutic response [Warnings and Precautions (5.4); Drug Interactions (7)].
Approximately 7% of the population has reduced activity of the CYP2D6 isoenzyme of cytochrome P-450. These individuals are "poor metabolizers" of debrisoquine, dextromethorphan, tricyclic antidepressants, among other drugs. Based on a population PK analysis of Phase I studies in healthy subjects, concentrations of tramadol were approximately 20% higher in "poor metabolizers" versus "extensive metabolizers", while M1 concentrations were 40% lower. Concomitant therapy with inhibitors of CYP2D6 such as fluoxetine, paroxetine and quinidine could result in significant drug interactions. In vitro drug interaction studies in human liver microsomes indicate that inhibitors of CYP2D6 such as fluoxetine and its metabolite norfluoxetine, amitriptyline and quinidine inhibit the metabolism of tramadol to various degrees, suggesting that concomitant administration of these compounds could result in increases in tramadol concentrations and decreased concentrations of M1. The full pharmacological impact of these alterations in terms of either efficacy or safety is unknown. Concomitant use of serotonin re-uptake inhibitors and MAO inhibitors may enhance the risk of adverse events, including seizure and serotonin syndrome [see Warnings and Precautions (5.8) and Drug Interactions (7)].
Excretion
Tramadol metabolites are eliminated primarily by the kidneys. Approximately 30% of the dose is excreted in the urine as unchanged drug, whereas 60% of the dose is excreted as metabolites. The remainder is excreted either as unidentified or as unextractable metabolites.
Special Populations
Hepatic Impairment
Metabolism of tramadol and M1 is reduced in patients with severe hepatic impairment based on a study in patients with advanced cirrhosis of the liver, resulting in both a larger area under the concentration time curve for tramadol and longer tramadol and M1 elimination half-lives (13 hrs. for tramadol and 19 hrs. for M1). In patients with severe hepatic impairment, adjustment of the dosing regimen is recommended [see Dosage and Administration (2)].
Renal Impairment
Impaired renal function results in a decreased rate and extent of excretion of tramadol and its active metabolite, M1. In patients with creatinine clearances of less than 30 mL/min, adjustment of the dosing regimen is recommended [see Dosage and Administration (2)]. The total amount of tramadol and M1 removed during a 4-hour dialysis period is less than 7% of the administered dose.
Age: Geriatric
Healthy elderly subjects aged 65 to 75 years have plasma tramadol concentrations and elimination half-lives comparable to those observed in healthy subjects less than 65 years of age. In subjects over 75 years, maximum serum concentrations are elevated (208 vs. 162 ng/mL) and the elimination half-life is prolonged (7 vs. 6 hours) compared to subjects 65 to 75 years of age. Adjustment of the daily dose is recommended for patients older than 75 years [see Dosage and Administration (2.2)].
Sex
The absolute bioavailability of tramadol was 73% in males and 79% in females. The plasma clearance was 6.4 mL/min/kg in males and 5.7 mL/min/kg in females following a 100 mg IV dose of tramadol. Following a single oral dose, and after adjusting for body weight, females had a 12% higher peak tramadol concentration and a 35% higher area under the concentration-time curve compared to males. The clinical significance of this difference is unknown.
Poor / Extensive Metabolizers, CYP2D6
The formation of the active metabolite, M1, is mediated by CYP2D6, a polymorphic enzyme. Approximately 7% of the population has reduced activity of the CYP2D6 isoenzyme of cytochrome P450 metabolizing enzyme system. These individuals are "poor metabolizers" of debrisoquine, dextromethorphan and tricyclic antidepressants, among other drugs. Based on a population PK analysis of Phase 1 studies with IR tablets in healthy subjects, concentrations of tramadol were approximately 20% higher in "poor metabolizers" versus "extensive metabolizers", while M1 concentrations were 40% lower.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
A slight, but statistically significant, increase in two common murine tumors, pulmonary and hepatic, was observed in an NMRI mouse carcinogenicity study, particularly in aged mice. Mice were dosed orally up to 30 mg/kg in the drinking water (0.36 times the MRHD) for approximately two years, although the study was not done with the Maximum Tolerated Dose. This finding is not believed to suggest risk in humans. No evidence of carcinogenicity was noted in a rat 2-year carcinogenicity study testing oral doses of up to 30 mg/kg in the drinking water, 0.73 times the MRHD.
Mutagenesis
Tramadol was mutagenic in the presence of metabolic activation in the mouse lymphoma assay. Tramadol was not mutagenic in the in vitro bacterial reverse mutation assay using Salmonella and E. coli (Ames), the mouse lymphoma assay in the absence of metabolic activation, the in vitro chromosomal aberration assay, or the in vivo micronucleus assay in bone marrow.
Impairment of Fertility
No effects on fertility were observed for tramadol at oral dose levels up to 50 mg/kg in male rats and 75 mg/kg in female rats. These dosages are 1.2 and 1.8 times the maximum recommended human daily dose based on body surface area, respectively.
14. Clinical Studies
ULTRAM has been given in single oral doses of 50, 75 and 100 mg to patients with pain following surgical procedures and pain following oral surgery (extraction of impacted molars).
In single-dose models of pain following oral surgery, pain relief was demonstrated in some patients at doses of 50 mg and 75 mg. A dose of 100 mg ULTRAM tended to provide analgesia superior to codeine sulfate 60 mg, but it was not as effective as the combination of aspirin 650 mg with codeine phosphate 60 mg.
ULTRAM has been studied in three long-term controlled trials involving a total of 820 patients, with 530 patients receiving ULTRAM. Patients with a variety of chronic painful conditions were studied in double-blind trials of one to three months duration. Average daily doses of approximately 250 mg of ULTRAM in divided doses were generally comparable to five doses of acetaminophen 300 mg with codeine phosphate 30 mg (TYLENOL with Codeine #3) daily, five doses of aspirin 325 mg with codeine phosphate 30 mg daily, or two to three doses of acetaminophen 500 mg with oxycodone hydrochloride 5 mg (TYLOX) daily.
Titration Trials
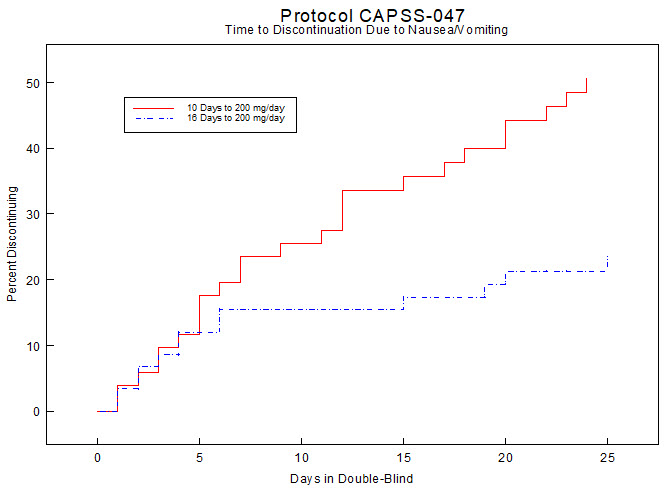
In a randomized, blinded clinical study with 129 to 132 patients per group, a 10-day titration to a daily ULTRAM dose of 200 mg (50 mg four times per day), attained in 50 mg increments every 3 days, was found to result in fewer discontinuations due to dizziness or vertigo than titration over only 4 days or no titration. In a second study with 54 to 59 patients per group, patients who had nausea or vomiting when titrated over 4 days were randomized to re-initiate ULTRAM therapy using slower titration rates.
A 16-day titration schedule, starting with 25 mg every morning and using additional doses in 25 mg increments every third day to 100 mg/day (25 mg four times per day), followed by 50 mg increments in the total daily dose every third day to 200 mg/day (50 mg four times per day), resulted in fewer discontinuations due to nausea or vomiting and fewer discontinuations due to any cause than did a 10-day titration schedule.
Figure 2:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.