XELJANZ Film-coated tablet Ref.[8636] Active ingredients: Tofacitinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
Pharmacodynamic properties
Pharmacotherapeutic groups: Immunosuppressants, Selective Immunosuppressants
ATC code: L04AA29
Mechanism of action
Tofacitinib is a potent, selective inhibitor of the JAK family. In enzymatic assays, tofacitinib inhibits JAK1, JAK2, JAK3, and to a lesser extent TyK2. In contrast, tofacitinib has a high degree of selectivity against other kinases in the human genome. In human cells, tofacitinib preferentially inhibits signalling by heterodimeric cytokine receptors that associate with JAK3 and/or JAK1 with functional selectivity over cytokine receptors that signal via pairs of JAK2. Inhibition of JAK1 and JAK3 by tofacitinib attenuates signalling of interleukins (IL-2, -4, -6, -7, -9, -15, -21) and type I and type II interferons, which will result in modulation of the immune and inflammatory response.
Pharmacodynamic effects
In patients with RA, treatment up to 6 months with tofacitinib was associated with dose-dependent reductions of circulating CD16/56+ natural killer (NK) cells, with estimated maximum reductions occurring at approximately 8-10 weeks after initiation of therapy. These changes generally resolved within 2-6 weeks after discontinuation of treatment. Treatment with tofacitinib was associated with dose-dependent increases in B cell counts. Changes in circulating T-lymphocyte counts and T-lymphocyte subsets (CD3+, CD4+ and CD8+) were small and inconsistent.
Following long-term treatment (median duration of tofacitinib treatment of approximately 5 years), CD4+ and CD8+ counts showed median reductions of 28% and 27%, respectively, from baseline. In contrast to the observed decrease after short-term dosing, CD16/56+ natural killer cell counts showed a median increase of 73% from baseline. CD19+ B cell counts showed no further increases after long-term tofacitinib treatment. All these lymphocyte subset changes returned toward baseline after temporary discontinuation of treatment. There was no evidence of a relationship between serious or opportunistic infections or herpes zoster and lymphocyte subset counts (see section 4.2 for absolute lymphocyte count monitoring).
Changes in total serum IgG, IgM, and IgA levels over 6-month tofacitinib dosing in patients with RA were small, not dose-dependent and similar to those seen on placebo, indicating a lack of systemic humoral suppression.
After treatment with tofacitinib in RA patients, rapid decreases in serum C-reactive protein (CRP) were observed and maintained throughout dosing. Changes in CRP observed with tofacitinib treatment do not reverse fully within 2 weeks after discontinuation, indicating a longer duration of pharmacodynamic activity compared to the half-life.
Vaccine studies
In a controlled clinical trial of patients with RA initiating tofacitinib 10 mg twice daily or placebo, the number of responders to influenza vaccine was similar in both groups: tofacitinib (57%) and placebo (62%). For pneumococcal polysaccharide vaccine the number of responders was as follows: 32% in patients receiving both tofacitinib and MTX; 62% for tofacitinib monotherapy; 62% for MTX monotherapy; and 77% for placebo. The clinical significance of this is unknown, however, similar results were obtained in a separate vaccine study with influenza and pneumococcal polysaccharide vaccines in patients receiving long-term tofacitinib 10 mg twice daily.
A controlled study was conducted in patients with RA on background MTX immunised with a live attenuated virus vaccine (Zostavax) 2 to 3 weeks before initiating a 12-week treatment with tofacitinib 5 mg twice daily or placebo. Evidence of humoral and cell-mediated responses to VZV was observed in both tofacitinib and placebo-treated patients at 6 weeks. These responses were similar to those observed in healthy volunteers aged 50 years and older. A patient with no previous history of varicella infection and no anti-varicella antibodies at baseline experienced dissemination of the vaccine strain of varicella 16 days after vaccination. Tofacitinib was discontinued and the patient recovered after treatment with standard doses of antiviral medication. This patient subsequently made a robust, though delayed, humoral and cellular response to the vaccine (see section 4.4).
Clinical efficacy and safety
Rheumatoid arthritis
The efficacy and safety of tofacitinib were assessed in 6 randomised, double-blind, controlled multicentre studies in patients greater than 18 years of age with active RA diagnosed according to American College of Rheumatology (ACR) criteria. Table 9 provides information regarding the pertinent study design and population characteristics.
Table 9. Phase 3 clinical trials of tofacitinib 5 mg and 10 mg twice daily doses in patients with RA:
| Studies | Study I (ORAL Solo) | Study II (ORAL Sync) | Study III (ORAL Standard) | Study IV (ORAL Scan) | Study V (ORAL Step) | Study VI (ORAL Start) | Study VII (ORAL Strategy) |
|---|---|---|---|---|---|---|---|
| Population | DMARD-IR | DMARD-IR | MTX-IR | MTX-IR | TNFi-IR | MTX-naïvea | MTX-IR |
| Control | Placebo | Placebo | Placebo | Placebo | Placebo | MTX | MTX, ADA |
| Background treatment | Noneb | csDMARD | MTX | MTX | MTX | Noneb | 3 Parallel arms: • Tofacitinib monotherapy • Tofacitinib+MTX • ADA+MTX |
| Key features | Monotherapy | Various csDMARDs | Active control (ADA) | X-Ray | TNFi-IR | Monotherapy, Active comparator (MTX), X-Ray | Tofacitinib with and without MTX in comparison to ADA with MTX |
| Number of patients treated | 610 | 792 | 717 | 797 | 399 | 956 | 1.146 |
| Total study duration | 6 months | 1 year | 1 year | 2 years | 6 months | 2 years | 1 year |
| Co-primary efficacy endpointsc | Month 3: ACR20 HAQ-DI DAS28-4(ESR)<2.6 | Month 6: ACR20 DAS28-4(ESR)<2.6 Month 3: HAQ-DI | Month 6: ACR20 DAS28-4(ESR)<2.6 Month 3: HAQ-DI | Month 6: ACR20 mTSS DAS28-4(ESR)<2.6 Month 3: HAQ-DI | Month 3: ACR20 HAQ-DI DAS28-4(ESR)<2.6 | Month 6: mTSS ACR70 | Month 6: ACR50 |
| Time of mandatory placebo rescue to tofacitinib 5 or 10 mg twice daily | Month 3 | Month 6 (placebo subjects with <20% improvement in swollen and tender joint counts advanced to tofacitinib at month 3) | Month 3 | NA | NA | ||
a ≤3 weekly doses (MTX-naïve).
b Antimalarials were allowed.
c Co-primary endpoints as follows: mean change from baseline in mTSS; percent of subjects achieving ACR20 or ACR70 responses; mean change from baseline in HAQ-DI; percent of subjects achieving a DAS28-4(ESR) <2.6 (remission).
mTSS=modified Total Sharp Score, ACR20(70)=American College of Rheumatology ≥20% (≥70%) improvement, DAS28=Disease Activity Score 28 joints, ESR=Erythrocyte Sedimentation Rate, HAQ-DI=Health Assessment Questionnaire Disability Index, DMARD=disease-modifying antirheumatic drug, IR=inadequate responder, csDMARD=conventional synthetic DMARD, TNFi=tumour necrosis factor inhibitor, NA=not applicable, ADA=adalimumab, MTX=methotrexate.
Clinical response
ACR response:
The percentages of tofacitinib-treated patients achieving ACR20, ACR50 and ACR70 responses in studies ORAL Solo, ORAL Sync, ORAL Standard, ORAL Scan, ORAL Step, ORAL Start, and ORAL Strategy are shown in Table 8. In all studies, patients treated with either 5 mg or 10 mg twice daily tofacitinib had statistically significant ACR20, ACR50 and ACR70 response rates at month 3 and month 6 versus placebo (or versus MTX in ORAL Start) treated patients.
Over the course of ORAL Strategy, responses with tofacitinib 5 mg twice daily + MTX were numerically similar compared to adalimumab 40 mg + MTX and both were numerically higher than tofacitinib 5 mg twice daily.
The treatment effect was similar in patients independent of rheumatoid fac tor status, age, gender, race, or disease status. Time to onset was rapid (as early as week 2 in studies ORAL Solo, ORAL Sync, and ORAL Step) and the magnitude of response continued to improve with duration of treatment. As with the overall ACR response in patients treated with 5 mg or 10 mg twice daily tofacitinib, each of the components of the ACR response was consistently improved from baseline including: tender and swollen joint counts; patient and physician global assessment; disability index scores; pain assessment and CRP compared to patients receiving placebo plus MTX or other DMARDs in all studies.
Table 10. Proportion (%) of patients with an ACR response:
| ORAL Solo: DMARD inadequate responders | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Endpoint | Time | Placebo N=122 | Tofacitinib 5 mg twice daily monotherapy N=241 | Tofacitinib 10 mg twice daily monotherapy N=243 | ||||||||||
| ACR20 | Month 3 | 26 | 60*** | 65*** | ||||||||||
| Month 6 | NA | 69 | 71 | |||||||||||
| ACR50 | Month 3 | 12 | 31*** | 37*** | ||||||||||
| Month 6 | NA | 42 | 47 | |||||||||||
| ACR70 | Month 3 | 6 | 15* | 20*** | ||||||||||
| Month 6 | NA | 22 | 29 | |||||||||||
| ORAL Sync: DMARD inadequate responders | ||||||||||||||
| Endpoint | Time | Placebo + DMARD(s) N=158 | Tofacitinib 5 mg twice daily + DMARD(s) N=312 | Tofacitinib 10 mg twice daily + DMARD(s) N=315 | ||||||||||
| ACR20 | Month 3 | 27 | 56*** | 63*** | ||||||||||
| Month 6 | 31 | 53*** | 57*** | |||||||||||
| Month 12 | NA | 51 | 56 | |||||||||||
| ACR50 | Month 3 | 9 | 27*** | 33*** | ||||||||||
| Month 6 | 13 | 34*** | 36*** | |||||||||||
| Month 12 | NA | 33 | 42 | |||||||||||
| ACR70 | Month 3 | 2 | 8** | 14*** | ||||||||||
| Month 6 | 3 | 13*** | 16*** | |||||||||||
| Month 12 | NA | 19 | 25 | |||||||||||
| ORAL Standard: MTX inadequate responders | ||||||||||||||
| Endpoint | Time | Placebo | Tofacitinib twice daily + MTX | Adalimumab 40 mg QOW + MTX | ||||||||||
| N=105 | 5 mg N=198 | 10 mg N=197 | N=199 | |||||||||||
| ACR20 | Month 3 | 26 | 59*** | 57*** | 56*** | |||||||||
| Month 6 | 28 | 51*** | 51*** | 46** | ||||||||||
| Month 12 | NA | 48 | 49 | 48 | ||||||||||
| ACR50 | Month 3 | 7 | 33*** | 27*** | 24*** | |||||||||
| Month 6 | 12 | 36*** | 34*** | 27** | ||||||||||
| Month 12 | NA | 36 | 36 | 33 | ||||||||||
| ACR70 | Month 3 | 2 | 12** | 15*** | 9* | |||||||||
| Month 6 | 2 | 19*** | 21*** | 9* | ||||||||||
| Month 12 | NA | 22 | 23 | 17 | ||||||||||
| ORAL Scan: MTX inadequate responders | ||||||||||||||
| Endpoint | Time | Placebo + MTX N=156 | Tofacitinib 5 mg twice daily + MTX N=316 | Tofacitinib 10 mg twice daily + MTX N=309 | ||||||||||
| ACR20 | Month 3 | 27 | 55*** | 66*** | ||||||||||
| Month 6 | 25 | 50*** | 62*** | |||||||||||
| Month 12 | NA | 47 | 55 | |||||||||||
| Month 24 | NA | 40 | 50 | |||||||||||
| ACR50 | Month 3 | 8 | 28*** | 36*** | ||||||||||
| Month 6 | 8 | 32*** | 44*** | |||||||||||
| Month 12 | NA | 32 | 39 | |||||||||||
| Month 24 | NA | 28 | 40 | |||||||||||
| ACR70 | Month 3 | 3 | 10** | 17*** | ||||||||||
| Month 6 | 1 | 14*** | 22*** | |||||||||||
| Month 12 | NA | 18 | 27 | |||||||||||
| Month 24 | NA | 17 | 26 | |||||||||||
| ORAL Step: TNF Inhibitor inadequate responders | ||||||||||||||
| Endpoint | Time | Placebo + MTX N=132 | Tofacitinib 5 mg twice daily + MTX N=133 | Tofacitinib 10 mg twice daily + MTX N=134 | ||||||||||
| ACR20 | Month 3 | 24 | 41* | 48*** | ||||||||||
| Month 6 | NA | 51 | 54 | |||||||||||
| ACR50 | Month 3 | 8 | 26*** | 28*** | ||||||||||
| Month 6 | NA | 37 | 30 | |||||||||||
| ACR70 | Month 3 | 2 | 14*** | 10* | ||||||||||
| Month 6 | NA | 16 | 16 | |||||||||||
| ORAL Start: MTX-naïve | ||||||||||||||
| Endpoint | Time | MTX N=184 | Tofacitinib 5 mg twice daily N=370 | Tofacitinib 10 mg twice daily N=394 | ||||||||||
| ACR20 | Month 3 | 52 | 69*** | 77*** | ||||||||||
| Month 6 | 51 | 71*** | 75*** | |||||||||||
| Month 12 | 51 | 67** | 71*** | |||||||||||
| Month 24 | 42 | 63*** | 64*** | |||||||||||
| ACR50 | Month 3 | 20 | 40*** | 49*** | ||||||||||
| Month 6 | 27 | 46*** | 56*** | |||||||||||
| Month 12 | 33 | 49** | 55*** | |||||||||||
| Month 24 | 28 | 48*** | 49*** | |||||||||||
| ACR70 | Month 3 | 5 | 20*** | 26*** | ||||||||||
| Month 6 | 12 | 25*** | 37*** | Month 12 | 15 | 28** | 38*** | Month 24 | 15 | 34*** | 37*** | |||
| ORAL Strategy: MTX inadequate responders | ||||||||||||||
| Endpoint | Time | Tofacitinib 5 mg twice daily N=384 | Tofacitinib 5 mg twice daily + MTX N=376 | Adalimumab + MTX N=386 | ||||||||||
| ACR20 | Month 3 | 62,50 | 70,48ǂ | 69,17 | ||||||||||
| Month 6 | 62,84 | 73,14ǂ | 70,98 | |||||||||||
| Month 12 | 61,72 | 70,21ǂ | 67,62 | |||||||||||
| ACR50 | Month 3 | 31,51 | 40,96ǂ | 37,31 | ||||||||||
| Month 6 | 38,28 | 46,01ǂ | 43,78 | |||||||||||
| Month 12 | 39,31 | 47,61ǂ | 45,85 | |||||||||||
| ACR70 | Month 3 | 13,54 | 19,41ǂ | 14,51 | ||||||||||
| Month 6 | 18,23 | 25,00ǂ | 20,73 | |||||||||||
| Month 12 | 21,09 | 28,99ǂ | 25,91 | |||||||||||
* p<0.05;
** p<0.001;
*** p<0.0001 verses placebo (versus MT X for ORAL Start);
ǂ p<0.05 – tofacitinib 5 mg + MT X versus tofacitinib 5 mg for ORAL Strategy (normal p-values without multiple comparison adjustment) QOW=every other week, N=number of subject s analysed, ACR20/50/70=American College of Rheumatology ≥20, 50, 70% improvement, NA=not applicable, MT X=methotrexate.
DAS28-4(ESR) response:
Patients in the phase 3 studies had a mean Disease Activity Score (DAS28-4[ESR]) of 6.1-6.7 at baseline. Significant reductions in DAS28-4(ESR) from baseline (mean improvement) of 1.8-2.0 and 1.9-2.2 were observed in patients treated with 5 mg and 10 mg twice daily doses, respectively, compared to placebo-treated patients (0.7-1.1) at month 3. The proportion of patients achieving a DAS28 clinical remission (DAS28-4(ESR) <2.6) in ORAL Step, ORAL Sync, and ORAL Standard is shown in Table 11.
Table 11. Number (%) of subjects achieving DAS28-4(ESR) <2.6 remission at months 3 and 6:
| Time Point | N | % | |
|---|---|---|---|
| ORAL Step: TNF Inhibitor inadequate responders | |||
| Tofacitinib 5 mg twice daily + MTX | Month 3 | 133 | 6 |
| Tofacitinib 10 mg twice daily + MTX | Month 3 | 134 | 8* |
| Placebo + MTX | Month 3 | 132 | 2 |
| ORAL Sync: DMARD inadequate responders | |||
| Tofacitinib 5 mg twice daily + MTX | Month 6 | 312 | 8* |
| Tofacitinib 10 mg twice daily + MTX | Month 6 | 315 | 11*** |
| Placebo | Month 6 | 158 | 3 |
| ORAL Standard: MTX inadequate responders | |||
| Tofacitinib 5 mg twice daily + MTX | Month 6 | 198 | 6* |
| Tofacitinib 10 mg twice daily + MTX | Month 6 | 197 | 11*** |
| Adalimumab 40 mg SC QOW + MTX | Month 6 | 199 | 6* |
| Placebo + MTX | Month 6 | 105 | 1 |
* p <0.05;
*** p <0.0001 versus placebo;
SC=subcutaneous, QOW=every other week, N=number of subjects analysed, DAS28=Disease Activity Scale 28 joints, ESR=Erythrocyte Sedimentation Rate.
Radiographic response:
In ORAL Scan and ORAL Start, inhibition of progression of structural joint damage was assessed radiographically and expressed as mean change from baseline in mTSS and its components, the erosion score and joint space narrowing (JSN) score, at months 6 and 12.
In ORAL Scan, tofacitinib 10 mg twice daily plus background MTX resulted in significantly greater inhibition of the progression of structural damage compared to placebo plus MTX at months 6 and 12. When given at a dose of 5 mg twice daily, tofacitinib plus MTX exhibited similar effects on mean progression of structural damage (not statistically significant). Analysis of erosion and JSN scores were consistent with overall results.
In the placebo plus MTX group, 78% of patients experienced no radiographic progression (mTSS change less than or equal to 0.5) at month 6 compared to 89% and 87% of patients treated with tofacitinib 5 or 10 mg (plus MTX) twice daily respectively, (both significant versus placebo plus MTX).
In ORAL Start, tofacitinib monotherapy resulted in significantly greater inhibition of the progression of structural damage compared to MTX at months 6 and 12 as shown in Table 10, which was also maintained at month 24. Analyses of erosion and JSN scores were consistent with overall results.
In the MTX group, 70% of patients experienced no radiographic progression at month 6 compared to 83% and 90% of patients treated with tofacitinib 5 or 10 mg twice daily respectively, both significant versus MTX.
Table 12. Radiographic changes at months 6 and 12:
| ORAL Scan: MTX inadequate responders | |||||
|---|---|---|---|---|---|
| Placebo + MTX N=139 Mean (SD)a | Tofacitinib 5 mg twice daily + MTX N=277 Mean (SD)a | Tofacitinib 5 mg twice daily + MTX Mean difference from placebob (CI) | Tofacitinib 10 mg twice daily + MTX N=290 Mean (SD)a | Tofacitinib 10 mg twice daily + MTX Mean difference from placebob (CI) | |
| mTSSc | |||||
| Baseline | 33 (42) | 31 (48) | - | 37 (54) | - |
| Month 6 | 0,5 (2,0) | 0,1 (1,7) | -0,3 (-0,7, 0,0) | 0,1 (2,0) | -0,4 (-0,8, 0,0) |
| Month 12 | 1,0 (3,9) | 0,3 (3,0) | -0,6 (-1,3, 0,0) | 0,1 (2,9) | -0,9 (-1,5, -0,2) |
| ORAL Start: MTX-naïve | |||||
| MTX N=168 Mean (SD)a | Tofacitinib 5 mg twice daily N=344 Mean (SD)a | Tofacitinib 5 mg twice daily Mean difference from MTXd (CI) | Tofacitinib 10 mg twice daily N=368 Mean (SD)a | Tofacitinib 10 mg twice daily Mean difference from MTXd (CI) | |
| mTSSc | |||||
| Baseline | 16 (29) | 20 (41) | - | 19 (39) | - |
| Month 6 | 0,9 (2,7) | 0,2 (2,3) | -0,7 (-1,0, -0,3) | 0,0 (1,2) | -0,8 (-1,2, -0,4) |
| Month 12 | 1,3 (3,7) | 0,4 (3,0) | -0,9 (-1,4, -0,4) | 0,0 (1,5) | -1,3 (-1,8, -0,8) |
a SD = Standard Deviat ion
b Difference between least squares means tofacitinib minus placebo (95% CI = 95% confidence interval)
c Month 6 and month 12 data are mean change from baseline
d Difference between least squares means tofacitinib minus MT X (95% CI = 95% confidence interval)
Physical function response and health-related outcomes:
Tofacitinib, alone or in combination with MTX, has shown improvements in physical function, as measured by the HAQ-DI. Patients receiving tofacitinib 5 or 10 mg twice daily demonstrated significantly greater improvement from baseline in physical functioning compared to placebo at month 3 (studies ORAL Solo, ORAL Sync, ORAL Standard, and ORAL Step) and month 6 (studies ORAL Sync and ORAL Standard). Tofacitinib 5 or 10 mg twice daily-treated patients demonstrated significantly greater improvement in physical functioning compared to placebo as early as week 2 in ORAL Solo and ORAL Sync. Changes from baseline in HAQ-DI in studies ORAL Standard, ORAL Step and ORAL Sync are shown in Table 13.
Table 13. LS mean change from baseline in HAQ-DI at month 3:
| Placebo + MTX | Tofacitinib 5 mg twice daily + MTX | Tofacitinib 10 mg twice daily + MTX | Adalimumab 40 mg QOW + MTX |
|---|---|---|---|
| ORAL Standard: MTX inadequate responders | |||
| N=96 | N=185 | N=183 | N=188 |
| -0,24 | -0,54*** | -0,61*** | -0,50*** |
| ORAL Step: TNF inhibitor inadequate responders | |||
| N=118 | N=117 | N=125 | NA |
| -0,18 | -0,43*** | -0,46*** | NA |
| Placebo + DMARD(s) | Tofacitinib 5 mg twice daily + DMARD(s) | Tofacitinib 10 mg twice daily + DMARD(s) | |
| ORAL Sync: DMARD inadequate responders | |||
| N=147 | N=292 | N=292 | NA |
| -0,21 | -0,46*** | -0,56*** | NA |
*** p<0.0001, tofacitinib versus placebo + MT X, LS = least squares, N = number of patients, QOW = every other week,
NA = not applicable, HAQ-DI = Health Assessment Questionnaire Disability Index
Health-related quality of life was assessed by the Short Form Health Survey (SF-36). Patients receiving either 5 or 10 mg tofacitinib twice daily experienced significantly greater improvement from baseline compared to placebo in all 8 domains as well as the Physical Component Summary and Mental Component Summary scores at month 3 in ORAL Solo, ORAL Scan and ORAL Step. In ORAL Scan, mean SF-36 improvements were maintained to 12 months in tofacitinib-treated patients.
Improvement in fatigue was evaluated by the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) scale at month 3 in all studies. Patients receiving tofacitinib 5 or 10 mg twice daily demonstrated significantly greater improvement from baseline in fatigue compared to placebo in all 5 studies. In ORAL Standard and ORAL Scan, mean FACIT-F improvements were maintained to 12 months in tofacitinib-treated patients.
Improvement in sleep was assessed using the Sleep Problems Index I and II summary scales of the Medical Outcomes Study Sleep (MOS-Sleep) measure at month 3 in all studies. Patients receiving tofacitinib 5 or 10 mg twice daily demonstrated significantly greater improvement from baseline in both scales compared to placebo in ORAL Sync, ORAL Standard and ORAL Scan. In ORAL Standard and ORAL Scan, mean improvements in both scales were maintained to 12 months in tofacitinib-treated patients.
Durability of clinical responses
Durability of effect was assessed by ACR20, ACR50, ACR70 response rates in studies of duration of up to two years. Changes in mean HAQ-DI and DAS28-4(ESR) were maintained in both tofacitinib treatment groups through to the end of the studies.
Evidence of persistence of efficacy with tofacitinib treatment for up to 5 years is also provided from data in a randomised post-authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, as well as in completed open-label, long-term follow-up studies up to 8 years.
Long-term controlled safety data
Study ORAL Surveillance (A3921133) was a large (N=4362), randomised active-controlled post-authorisation safety surveillance study of rheumatoid arthritis patients who were 50 years of age and older and had at least one additional cardiovascular risk factor (CV risk factors defined as: current cigarette smoker, diagnosis of hypertension, diabetes mellitus, family history of premature coronary heart disease, history of coronary artery disease including a history of revascularization procedure, coronary artery bypass grafting, myocardial infarction, cardiac arrest, unstable angina, acute coronary syndrome, and presence of extra-articular disease associated with RA, e.g. nodules, Sjögren's syndrome, anaemia of chronic disease, pulmonary manifestations). The majority (more than 90%) of tofacitinib patients who were current or past smokers had a smoking duration of more than 10 years and a median of 35.0 and 39.0 smoking years, respectively. Patients were required to be on a stable dose of methotrexate at study entry; dose adjustment was permitted during the study.
Patients were randomised to open-label tofacitinib 10 mg twice daily, tofacitinib 5 mg twice daily, or a TNF inhibitor (TNF inhibitor was either etanercept 50 mg once weekly or adalimumab 40 mg every other week) in a 1:1:1 ratio. The co-primary endpoints were adjudicated malignancies excluding NMSC and adjudicated major adverse cardiovascular events (MACE); cumulative incidence and statistical assessment of endpoints were blinded. The study was an event-powered study that also required at least 1500 patients to be followed for 3 years. The study treatment of tofacitinib 10 mg twice daily was stopped and patients were switched to 5 mg twice daily because of a dose-dependent signal of venous thromboembolic events (VTE). For patients in the tofacitinib 10 mg twice daily treatment arm, the data collected before and after the dose switch were analysed in their originally randomised treatment group.
The study did not meet the non-inferiority criterion for the primary comparison of the combined tofacitinib doses to TNF inhibitor since the upper limit of the 95% CI for HR exceeded the pre-specified non-inferiority criterion of 1.8 for adjudicated MACE and adjudicated malignancies excluding NMSC.
The results for adjudicated MACE, adjudicated malignancies excluding NMSC, and selected other events are provided below.
MACE (including myocardial infarction) and venous thromboembolism (VTE)
An increase in non-fatal myocardial infarction was observed in patients treated with tofacitinib compared to TNF inhibitor. A dose-dependent increase in VTE events was observed in patients treated with tofacitinib compared to TNF inhibitor (see sections 4.4 and 4.8).
Table 14. Incidence rate and hazard ratio for MACE, myocardial infarction and venous thromboembolism:
| Tofacitinib 5 mg twice daily | Tofacitinib 10 mg twice dailya | All Tofacitinibb | TNF inhibitor (TNFi) | |
|---|---|---|---|---|
| MACEc | ||||
| IR (95% CI) per 100 PY | 0,91 (0,67, 1,21) | 1,05 (0,78, 1,38) | 0,98 (0,79, 1,19) | 0,73 (0,52, 1,01) |
| HR (95% CI) vs TNFi | 1,24 (0,81, 1,91) | 1,43 (0,94, 2,18) | 1,33 (0,91, 1,94) | |
| Fatal MIc | ||||
| IR (95% CI) per 100 PY | 0,00 (0,00, 0,07) | 0,06 (0,01, 0,18) | 0,03 (0,01, 0,09) | 0,06 (0,01, 0,17) |
| HR (95% CI) vs TNFi | 0,00 (0,00, Inf) | 1,03 (0,21, 5,11) | 0,50 (0,10, 2,49) | |
| Non-fatal MIc | ||||
| IR (95% CI) per 100 PY | 0,37 (0,22, 0,57) | 0,33 (0,19, 0,53) | 0,35 (0,24, 0,48) | 0,16 (0,07, 0,31) |
| HR (95% CI) vs TNFi | 2,32 (1,02, 5,30) | 2,08 (0,89, 4,86) | 2,20 (1,02, 4,75) | |
| VTEd | ||||
| IR (95% CI) per 100 PY | 0,33 (0,19, 0,53) | 0,70 (0,49, 0,99) | 0,51 (0,38, 0,67) | 0,20 (0,10, 0,37) |
| HR (95% CI) vs TNFi | 1,66 (0,76, 3,63) | 3,52 (1,74, 7,12) | 2,56 (1,30, 5,05) | |
| PEd | ||||
| IR (95% CI) per 100 PY | 0,17 (0,08, 0,33) | 0,50 (0,32, 0,74) | 0,33 (0,23, 0,46) | 0,06 (0,01, 0,17) |
| HR (95% CI) vs TNFi | 2,93 (0,79, 10,83) | 8,26 (2,49, 27,43) | 5,53 (1,70, 18,02) | |
| DVTd | ||||
| IR (95% CI) per 100 PY | 0,21 (0,11, 0,38) | 0,31 (0,17, 0,51) | 0,26 (0,17, 0,38) | 0,14 (0,06, 0,29) |
| HR (95% CI) vs TNFi | 1,54 (0,60, 3,97) | 2,21 (0,90, 5,43) | 1,87 (0,81, 4,30) | |
a The tofacitinib 10 mg twice daily treatment group includes data from patients that were switched from tofacitinib 10 mg twice daily to tofacitinib 5 mg twice daily as a result of a study modification.
b Combined tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily.
c Based on events occurring on treatment or within 60 days of treatment discontinuation.
d Based on events occurring on treatment or within 28 days of treatment discontinuation.
Abbreviations: MACE = major adverse cardiovascular events, MI = myocardial infarction, VTE = venous thromboembolism, PE = pulmonary embolism, DVT = deep vein thrombosis, TNF = tumour necrosis factor, IR = incidence rate, HR = hazard ratio, CI = confidence interval, PY = patient years, Inf = infinity
The following predictive factors for development of MI (fatal and non-fatal) were identified using a multivariate Cox model with backward selection: age ≥65 years, male, current or past smoking, history of diabetes, and history of coronary artery disease (which includes myocardial infarction, coronary heart disease, stable angina pectoris, or coronary artery procedures) (see sections 4.4 and 4.8).
Malignancies
An increase in malignancies excluding NMSC, particularly lung cancer, lymphoma and an increase in NMSC was observed in patients treated with tofacitinib compared to TNF inhibitor.
Table 15. Incidence rate and hazard ratio for malignanciesa:
| Tofacitinib 5 mg twice daily | Tofacitinib 10 mg twice dailyb | All Tofacitinibc | TNF inhibitor (TNFi) | |
|---|---|---|---|---|
| Malignancies excluding NMSC | ||||
| IR (95% CI) per 100 PY | 1,13 (0,87, 1,45) | 1,13 (0,86, 1,45) | 1,13 (0,94, 1,35) | 0,77 (0,55, 1,04) |
| HR (95% CI) vs TNFi | 1,47 (1,00, 2,18) | 1,48 (1,00, 2,19) | 1,48 (1,04, 2,09) | |
| Lung cancer | ||||
| IR (95% CI) per 100 PY | 0,23 (0,12, 0,40) | 0,32 (0,18, 0,51) | 0,28 (0,19, 0,39) | 0,13 (0,05, 0,26) |
| HR (95% CI) vs TNFi | 1,84 (0,74, 4,62) | 2,50 (1,04, 6,02) | 2,17 (0,95, 4,93) | |
| Lymphoma | ||||
| IR (95% CI) per 100 PY | 0,07 (0,02, 0,18) | 0,11 (0,04, 0,24) | 0,09 (0,04, 0,17) | 0,02 (0,00, 0,10) |
| HR (95% CI) vs TNFi | 3,99 (0,45, 35,70) | 6,24 (0,75, 51,86) | 5,09 (0,65, 39,78) | |
| NMSC | ||||
| IR (95% CI) per 100 PY | 0,61 (0,41, 0,86) | 0,69 (0,47, 0,96) | 0,64 (0,50, 0,82) | 0,32 (0,18, 0,52) |
| HR (95% CI) vs TNFi | 1,90 (1,04, 3,47) | 2,16 (1,19, 3,92) | 2,02 (1,17, 3,50) | |
a For malignancies excluding NMSC, lung cancer, and lymphoma, based on events occurring on treatment or after treatment discontinuation up to the end of the study. For NMSC based on events occurring on treatment or within 28 days of treatment discontinuation.
b The tofacitinib 10 mg twice daily treatment group includes data from patients that were switched from tofacitinib 10 mg twice daily to tofacitinib 5 mg twice daily as a result of a study modification.
c Combined tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily.
Abbreviations: NMSC = non melanoma skin cancer, TNF = tumour necrosis factor, IR = incidence rate, HR = hazard ratio, CI = confidence interval, PY = patient years
The following predictive factors for development of malignancies excluding NMSC were identified using a Multivariate Cox model with backward selection: age ≥65 years and current or past smoking (see sections 4.4 and 4.8).
Mortality
Increased mortality was observed in patients treated with tofacitinib compared to TNF inhibitors. Mortality was mainly due to cardiovascular events, infections and malignancies.
Table 16. Incidence rate and hazard ratio for mortalitya:
| Tofacitinib 5 mg twice daily | Tofacitinib 10 mg twice dailyb | All Tofacitinibc | TNF inhibitor (TNFi) | |
|---|---|---|---|---|
| Mortality (all cause) | ||||
| IR (95% CI) per 100 PY | 0,50 (0,33, 0,74) | 0,80 (0,57, 1,09) | 0,65 (0,50, 0,82) | 0,34 (0,20, 0,54) |
| HR (95% CI) vs TNFi | 1,49 (0,81, 2,74) | 2,37 (1,34, 4,18) | 1,91 (1,12, 3,27) | |
| Fatal infections | ||||
| IR (95% CI) per 100 PY | 0,08 (0,02, 0,20) | 0,18 (0,08, 0,35) | 0,13 (0,07, 0,22) | 0,06 (0,01, 0,17) |
| HR (95% CI) vs TNFi | 1,30 (0,29, 5,79) | 3,10 (0,84, 11,45) | 2,17 (0,62, 7,62) | |
| Fatal CV events | ||||
| IR (95% CI) per 100 PY | 0,25 (0,13, 0,43) | 0,41 (0,25, 0,63) | 0,33 (0,23, 0,46) | 0,20 (0,10, 0,36) |
| HR (95% CI) vs TNFi | 1,26 (0,55, 2,88) | 2,05 (0,96, 4,39) | 1,65 (0,81, 3,34) | |
| Fatal Malignancies | ||||
| IR (95% CI) per 100 PY | 0,10 (0,03, 0,23) | 0,00 (0,00, 0,08) | 0,05 (0,02, 0,12) | 0,02 (0,00, 0,11) |
| HR (95% CI) vs TNFi | 4,88 (0,57, 41,74) | 0 (0,00, Inf) | 2,53 (0,30, 21,64) | |
a Based on events occurring on treatment or within 28 days of treatment discontinuation.
b The tofacitinib 10 mg twice daily treatment group includes data from patients that were switched from tofacitinib 10 mg twice daily to tofacitinib 5 mg twice daily as a result of a study modification.
c Combined tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily.
Abbreviations: TNF = tumor necrosis factor, IR = incidence rate, HR = hazard ratio, CI = confidence interval, PY = patient years, CV = cardiovascular, Inf = infinity
Psoriatic arthritis
The efficacy and safety of tofacitinib film-coated tablets were assessed in 2 randomised, double-blind, placebo-controlled Phase 3 studies in adult patients with active PsA (≥ 3 swollen and ≥ 3 tender joints). Patients were required to have active plaque psoriasis at the screening visit. For both studies, the primary endpoints were ACR20 response rate and change from baseline in HAQ-DI at month 3.
Study PsA-I (OPAL BROADEN) evaluated 422 patients who had a previous inadequate response (due to lack of efficacy or intolerance) to a csDMARD (MTX for 92.7% of patients); 32.7% of the patients in this study had a previous inadequate response to > 1 csDMARD or 1 csDMARD and a targeted synthetic DMARD (tsDMARD). In OPAL BROADEN, previous treatment with TNF inhibitor was not allowed. All patients were required to have 1 concomitant csDMARD; 83.9% of patients received concomitant MTX, 9.5% of patients received concomitant sulfasalazine, and 5.7% of patients received concomitant leflunomide. The median PsA disease duration was 3.8 years. At baseline, 79.9% and 56.2% of patients had enthesitis and dactylitis, respectively. Patients randomised to tofacitinib received 5 mg twice daily or tofacitinib 10 mg twice daily for 12 months. Patients randomised to placebo were advanced in a blinded manner at month 3 to either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily and received treatment until month 12. Patients randomised to adalimumab (active-control arm) received 40 mg subcutaneously every 2 weeks for 12 months.
Study PsA-II (OPAL BEYOND) evaluated 394 patients who had discontinued a TNF inhibitor due to lack of efficacy or intolerance; 36.0% had a previous inadequate response to > 1 biological DMARD. All patients were required to have 1 concomitant csDMARD; 71.6% of patients received concomitant MTX, 15.7% of patients received concomitant sulfasalazine, and 8.6% of patients received concomitant leflunomide. The median PsA disease duration was 7.5 years. At baseline, 80.7% and 49.2% of patients had enthesitis and dactylitis, respectively. Patients randomised to tofacitinib received 5 mg twice daily or tofacitinib 10 mg twice daily for 6 months. Patients randomised to placebo were advanced in a blinded manner at month 3 to either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily and received treatment until month 6.
Signs and symptoms
Treatment with tofacitinib resulted in significant improvements in some signs and symptoms of PsA, as assessed by the ACR20 response criteria compared to placebo at month 3. The efficacy results for important endpoints assessed are shown in Table 17.
Table 17. Proportion (%) of PsA patients who achieved clinical response and mean change from baseline in OPAL BROADEN and OPAL BEYOND studies:
| Conventional synthetic DMARD inadequate respondersa (TNFi-Naïve) | TNFi inadequate respondersb | ||||
|---|---|---|---|---|---|
| OPAL BROADEN | OPAL BEYONDc | ||||
| Treatment group | Placebo | Tofacitinib 5 mg twice daily | Adalimumab 40 mg SC q2W | Placebo | Tofacitinib 5 mg twice daily |
| N | 105 | 107 | 106 | 131 | 131 |
| ACR20 | |||||
| Month 3 | 33% | 50%d* | 52%* | 24% | 50%d*** |
| Month 6 | NA | 59% | 64% | NA | 60% |
| Month 12 | NA | 68% | 60% | - | - |
| ACR50 | |||||
| Month 3 | 10% | 28%e** | 33%*** | 15% | 30%e* |
| Month 6 | NA | 38% | 42% | NA | 38% |
| Month 12 | NA | 45% | 41% | - | - |
| ACR70 | |||||
| Month 3 | 5% | 17%e* | 19%* | 10% | 17% |
| Month 6 | NA | 18% | 30% | NA | 21% |
| Month 12 | NA | 23% | 29% | - | - |
| ∆LEIf | |||||
| Month 3 | -0,4 | -0,8 | -1,1* | -0,5 | -1,3* |
| Month 6 | NA | -1,3 | -1,3 | NA | -1,5 |
| Month 12 | NA | -1,7 | -1,6 | - | - |
| ∆DSSf | |||||
| Month 3 | -2,0 | -3,5 | -4,0 | -1,9 | -5,2* |
| Month 6 | NA | -5,2 | -5,4 | NA | -6,0 |
| Month 12 | NA | -7,4 | -6,1 | - | - |
| PASI75g | |||||
| Month 3 | 15% | 43%d*** | 39%** | 14% | 21% |
| Month 6 | NA | 46% | 55% | NA | 34% |
| Month 12 | NA | 56% | 56% | - | - |
* Nominal p≤0.05;
** Nominal p<0.001;
*** Nominal p<0.0001 for active treatment versus placebo at month 3.
Abbreviations: BSA=body surface area; ∆LEI=change from baseline in Leeds Enthesitis Index; ∆DSS=change from baseline in Dactylitis Severity Score; ACR20/50/70=American College of Rheumatology ≥20%, 50%, 70% improvement; csDMARD=conventional synthetic disease-modifying antirheumatic drug; N=number of randomised and treated patients; NA=Not applicable, as data for placebo treatment is not available beyond month 3 due to placebo advanced to tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily; SC q2w=subcutaneously once every 2 weeks; TNFi=tumour necrosis factor inhibitor; PASI=Psoriasis Area and Severity index; PASI75=≥ 75% improvement in PASI.
a Inadequate response to at least 1 csDMARD due to lack of efficacy and/or intolerability.
b Inadequate response to a least 1 TNFi due to lack of efficacy and/or intolerability.
c OPAL BEYOND had a duration of 6 months.
d Achieved statistical significance globally at p≤ 0.05 per the pre-specified step-down testing procedure.
e Achieved statistical significance within the ACR family (ACR50 and ACR70) at p≤ 0.05 per the pre-specified step-down testing procedure.
f For patients with Baseline score >0.
g For patients with Baseline BSA ≥3% and PASI >0.
Both TNF inhibitor naïve and TNF inhibitor inadequate responder tofacitinib 5 mg twice daily -treated patients had significantly higher ACR20 response rates compared to placebo at month 3. Examination of age, sex, race, baseline disease activity and PsA subtype did not identify differences in response to tofacitinib. The number of patients with arthritis mutilans or axial involvement was too small to allow meaningful assessment. Statistically significant ACR20 response rates were observed with tofacitinib 5 mg twice daily in both studies as early as week 2 (first post-baseline assessment) in comparison to placebo.
In OPAL BROADEN, Minimal Disease Activity (MDA) response was achieved by 26.2%, 25.5% and 6.7% of tofacitinib 5 mg twice daily, adalimumab and placebo treated patients, respectively (tofacitinib 5 mg twice daily treatment difference from placebo 19.5% [95% CI: 9.9, 29.1]) at month 3. In OPAL BEYOND, MDA was achieved by 22.9% and 14.5% of tofacitinib 5 mg twice daily and placebo treated patients, respectively, however tofacitinib 5 mg twice daily did not achieve nominal statistical significance (treatment difference from placebo 8.4% [95% CI: -1.0, 17.8] at month 3).
Radiographic response
In study OPAL BROADEN, the progression of structural joint damage was assessed radiographically utilising the van der Heijde modified Total Sharp Score (mTSS) and the proportion of patients with radiographic progression (mTSS increase from baseline greater than 0.5) was assessed at month 12. At month 12, 96% and 98% of patients receiving tofacitinib 5 mg twice daily, and adalimumab 40 mg subcutaneously every 2 weeks, respectively, did not have radiographic progression (mTSS increase from baseline less than or equal to 0.5).
Physical function and health-related quality of life
Improvement in physical functioning was measured by the HAQ-DI. Patients receiving tofacitinib 5 mg twice daily demonstrated greater improvement (p≤ 0.05) from baseline in physical functioning compared to placebo at month 3 (see Table 18).
Table 18. Change from baseline in HAQ-DI in PsA studies OPAL BROADEN and OPAL BEYOND:
| Least squares mean change from baseline in HAQ-DI | |||||
|---|---|---|---|---|---|
| Conventional synthetic DMARD inadequate respondersa (TNFi-naïve) | TNFi inadequate respondersb | ||||
| OPAL BROADEN | OPAL BEYOND | ||||
| Treatment group | Placebo | Tofacitinib 5 mg twice daily | Adalimumab 40 mg SC q2W | Placebo | Tofacitinib 5 mg twice daily |
| N | 104 | 107 | 106 | 131 | 129 |
| Month 3 | -0,18 | -0,35c* | -0,38* | -0,14 | -0,39c*** |
| Month 6 | NA | -0,45 | -0,43 | NA | -0,44 |
| Month 12 | NA | -0,54 | -0,45 | NA | NA |
* Nominal p≤0.05;
*** Nominal p<0.0001 for active treatment versus placebo at month 3.
Abbreviations: DMARD=disease-modifying antirheumatic drug; HAQ-DI=Health Assessment Questionnaire Disability Index; N=total number of patients in the statistical analysis; SC q2w=subcutaneously once every 2 weeks; TNFi=tumour necrosis factor inhibitor.
a Inadequate response to at least one conventional synthetic DMARD (csDMARD) due to lack of efficacy and/or intolerability.
b Inadequate response to a least one TNF inhibitor (TNFi) due to lack of efficacy and/or intolerability.
c Achieved statistical significance globally at p≤ 0.05 per the pre-specified step-down testing procedure.
The HAQ-DI responder rate (response defined as having decrease from baseline of ≥ 0.35) at month 3 in studies OPAL BROADEN and OPAL BEYOND was 53% and 50%, respectively in patients receiving tofacitinib 5 mg twice daily, 31% and 28%, respectively in patients receiving placebo, and 53% in patients receiving adalimumab 40 mg subcutaneously once every 2 weeks (OPAL BROADEN only).
Health-related quality of life was assessed by SF-36v2, fatigue was assessed by the FACIT-F. Patients receiving tofacitinib 5 mg twice daily demonstrated greater improvement from baseline compared to placebo in the SF-36v2 physical functioning domain, the SF-36v2 physical component summary score, and FACIT-F scores at month 3 in studies OPAL BROADEN and OPAL BEYOND (nominal p≤ 0.05). Improvements from baseline in SF-36v2 and FACIT-F were maintained through month 6 (OPAL BROADEN and OPAL BEYOND) and month 12 (OPAL BROADEN).
Patients receiving tofacitinib 5 mg twice daily demonstrated a greater improvement in arthritis pain (as measured on a 0-100 visual analogue scale) from baseline at week 2 (first post-baseline assessment) through month 3 compared to placebo in studies OPAL BROADEN and OPAL BEYOND (nominal p ≤0.05).
Ankylosing spondylitis
The tofacitinib clinical development program to assess the efficacy and safety included one placebo-controlled confirmatory trial (Study AS-I). Study AS-I was a randomised, double-blind, placebo-controlled, 48-week treatment clinical study in 269 adult patients who had an inadequate response (inadequate clinical response or intolerance) to at least 2 NSAIDs. Patients were randomised and treated with tofacitinib 5 mg twice daily or placebo for 16 weeks of blinded treatment and then all were advanced to tofacitinib 5 mg twice daily for an additional 32 weeks. Patients had active disease as defined by both Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) and back pain score (BASDAI question 2) of greater or equal to 4 despite non-steroidal anti-inflammatory drug (NSAID), corticosteroid or DMARD therapy.
Approximately 7% and 21% of patients used concomitant methotrexate or sulfasalazine, respectively, from baseline to Week 16. Patients were allowed to receive a stable low dose of oral corticosteroids (8.6% received) and/or NSAIDs (81.8% received) from baseline to Week 48. Twenty-two percent of patients had an inadequate response to 1 or 2 TNF blockers. The primary endpoint was to evaluate the proportion of patients who achieved an ASAS20 response at Week 16.
Clinical response
Patients treated with tofacitinib 5 mg twice daily achieved greater improvements in ASAS20 and ASAS40 responses compared to placebo at Week 16 (Table 19). The responses were maintained from Week 16 through to Week 48 in patients receiving tofacitinib 5 mg twice daily.
Table 19. ASAS20 and ASAS40 Responses at Week 16, Study AS-I:
| Placebo (N=136) | Tofacitinib 5 mg Twice Daily (N=133) | Difference from Placebo (95% CI) | |
|---|---|---|---|
| ASAS20 response*, % | 29 | 56 | 27 (16, 38)** |
| ASAS40 response*, % | 13 | 41 | 28 (18, 38)** |
* type I error-controlled.
** p <0.0001.
The efficacy of tofacitinib was demonstrated in bDMARD naïve and TNF-inadequate responders (IR)/bDMARD experienced (non-IR) patients (Table 20).
Table 20. ASAS20 and ASAS40 Responses (%) by Treatment History at Week 16, Study AS-I:
| Prior Treatment History | Efficacy Endpoint | |||||
| ASAS20 | ASAS40 | |||||
| Placebo N | Tofacitinib 5 mg Twice Daily N | Difference from Placebo (95% CI) | Placebo N | Tofacitinib 5 mg Twice Daily N | Difference from Placebo (95% CI) | |
| bDMARD-Naïve | 105 | 102 | 28 (15, 41) | 105 | 102 | 31 (19, 43) |
| TNFi-IR or bDMARD Use (Non-IR) | 31 | 31 | 23 (1, 44) | 31 | 31 | 19 (2, 37) |
ASAS20 = An improvement from Baseline ≥ 20% and ≥ 1 unit increase in at least 3 domains on a scale of 0 to 10, and no worsening of ≥ 20% and ≥ 1 unit in the remaining domain; ASAS40 = An improvement from Baseline ≥ 40% and ≥ 2 units in at least 3 domains on a scale of 0 to 10 and no worsening at all in the remaining domain; bDMARD = biologic disease-modifying anti-rheumatic drug; CI = confidence interval; Non-IR = non-inadequate response; TNFi-IR = tumour necrosis factor inhibitor inadequate response.
The improvements in the components of the ASAS response and other measures of disease activity were higher in tofacitinib 5 mg twice daily compared to placebo at Week 16 as shown in Table 21.
The improvements were maintained from Week 16 through to Week 48 in patients receiving tofacitinib 5 mg twice daily.
Table 21. ASAS Components and Other Measures of Disease Activity at Week 16, Study AS-I:
| Placebo (N=136) | Tofacitinib 5 mg Twice Daily (N=133) | ||||
| Baseline (mean) | Week 16 (LSM change from Baseline) | Baseline (mean) | Week 16 (LSM change from Baseline) | Difference from Placebo (95% CI) | |
| ASAS Components | |||||
| - Patient Global Assessment of Disease Activity (0-10)a,* | 7,0 | -0,9 | 6,9 | -2,5 | -1,6 (-2,07, -1,05)** |
| - Total spinal pain (0-10)a,* | 6,9 | -1,0 | 6,9 | -2,6 | -1,6 (-2,10, -1,14)** |
| BASFI (0-10)b,* | 5,9 | -0,8 | 5,8 | -2,0 | -1,2 (-1,66, -0,80)** |
| - Inflammation (0-10)c,* | 6,8 | -1.0 | 6,6 | -2,7 | -1,7 (-2,18, -1,25)** |
| - BASDAI Scored | 6,5 | -1,1 | 6,4 | -2,6 | -1,4 (-1,88, -1,00)** |
| BASMIe,* | 4,4 | -0,1 | 4,5 | -0,6 | -0,5 (-0,67, -0,37)** |
| hsCRPf,* (mg/dL) | 1,8 | -0,1 | 1,6 | -1,1 | -1,0 (-1,20, -0,72)** |
| ASDAScrpg,* | 3,9 | -0,4 | 3,8 | -1,4 | -1,0 (-1,16, -0,79)** |
* type I error-controlled.
** p < 0.0001.
a Measured on a numerical rating scale with 0 = not active or no pain, 10 = very active or most severe pain.
b Bath Ankylosing Spondylitis Functional Index measured on a numerical rating scale with 0 = easy and 10 = impossible.
c Inflammation is the mean of two patient-reported stiffness self-assessments in BASDAI.
d Bath Ankylosing Spondylitis Disease Activity Index total score.
e Bath Ankylosing Spondylitis Metrology Index.
f High sensitivity C-reactive protein.
g Ankylosing Spondylitis Disease Activity Score with C-reactive protein.
LSM = least squares mean
Other health-related outcomes
Patients treated with tofacitinib 5 mg twice daily achieved greater improvements from baseline in Ankylosing Spondylitis Quality of Life (ASQoL) (-4.0 vs -2.0) and Functional Assessment of Chronic Illness Therapy - Fatigue (FACIT-F) Total score (6.5 vs 3.1) compared to placebo-treated patients at Week 16 (p<0.001). Patients treated with tofacitinib 5 mg twice daily achieved consistently greater improvements from baseline in the Short Form health survey version 2 (SF-36v2), Physical Component Summary (PCS) domain compared to placebo-treated patients at Week 16.
Ulcerative colitis
The efficacy and safety of tofacitinib film-coated tablets for the treatment of adult patients with moderately to severely active UC (Mayo score 6 to 12 with endoscopy subscore ≥ 2 and rectal bleeding subscore ≥ 1) were assessed in 3 multicentre, double-blind, randomised, placebo-controlled studies: 2 identical induction studies (OCTAVE Induction 1 and OCTAVE Induction 2) followed by 1 maintenance study (OCTAVE Sustain). Enrolled patients had failed at least 1 conventional therapy, including corticosteroids, immunomodulators, and/or a TNF inhibitor. Concomitant stable doses of oral aminosalicylates and corticosteroids (prednisone or equivalent daily dose up to 25 mg) were permitted with taper of corticosteroids to discontinuation mandated within 15 weeks of entering the maintenance study. Tofacitinib was administered as monotherapy (i.e., without concomitant use of biologics and immunosuppressants) for UC.
Table 22 provides additional information regarding pertinent study design and population characteristics.
Table 22. Phase 3 clinical studies of tofacitinib 5 mg and 10 mg twice daily doses in patients with UC:
| OCTAVE Induction 1 | OCTAVE Induction 2 | OCTAVE Sustain | |
|---|---|---|---|
| Treatment groups (randomisation ratio) | Tofacitinib 10 mg twice daily placebo (4:1) | Tofacitinib 10 mg twice daily placebo (4:1) | Tofacitinib 5 mg twice daily Tofacitinib 10 mg twice daily placebo (1:1:1) |
| Number of patients enrolled | 598 | 541 | 593 |
| Study duration | 8 weeks | 8 weeks | 52 weeks |
| Primary efficacy endpoint | Remission | Remission | Remission |
| Key secondary efficacy endpoints | Improvement of endoscopic appearance of the mucosa | Improvement of endoscopic appearance of the mucosa | Improvement of endoscopic appearance of the mucosa. Sustained corticosteroid-free remission among patients in remission at baseline |
| Prior TNFi failure | 51,3% | 52,1% | 44,7% |
| Prior corticosteroid failure | 74,9% | 71,3% | 75,0% |
| Prior immunosuppressant failure | 74,1% | 69,5% | 69,6% |
| Baseline corticosteroid use | 45,5% | 46,8% | 50,3% |
Abbreviations: TNFi=tumour necrosis factor inhibitor; UC=ulcerative colitis.
In addition, safety and efficacy of tofacitinib were assessed in an open-label long-term extension study (OCTAVE Open). Patients who completed 1 of the induction studies (OCTAVE Induction 1 or OCTAVE Induction 2) but did not achieve clinical response or patients who completed or withdrew early due to treatment failure in the maintenance study (OCTAVE Sustain) were eligible for OCTAVE Open. Patients from OCTAVE Induction 1 or OCTAVE Induction 2 who did not achieve clinical response after 8 weeks in OCTAVE Open were to be discontinued from OCTAVE Open. Corticosteroid tapering was also required upon entrance into OCTAVE Open.
Induction efficacy data (OCTAVE Induction 1 and OCTAVE Induction 2)
The primary endpoint of OCTAVE Induction 1 and OCTAVE Induction 2 was the proportion of patients in remission at week 8, and the key secondary endpoint was the proportion of patients with improvement of endoscopic appearance of the mucosa at week 8. Remission was defined as clinical remission (a total Mayo score ≤ 2 with no individual subscore > 1) and rectal bleeding subscore of 0. Improvement of endoscopic appearance of the mucosa was defined as endoscopy subscore of 0 or 1.
A significantly greater proportion of patients treated with tofacitinib 10 mg twice daily achieved remission, improvement of endoscopic appearance of the mucosa, and clinical response at week 8 compared to placebo in both studies, as shown in Table 23.
The efficacy results based on the endoscopic readings at the study sites were consistent with the results based on the central endoscopy readings.
Table 23. Proportion of patients meeting efficacy endpoints at week 8 (OCTAVE induction study 1 and OCTAVE induction study 2):
| OCTAVE induction study 1 | ||||
|---|---|---|---|---|
| Central endoscopy read | Local endoscopy read | |||
| Endpoint | Placebo | Tofacitinib 10 mg twice daily | Placebo | Tofacitinib 10 mg twice daily |
| N=122 | N=476 | N=122 | N=476 | |
| Remissiona | 8,2% | 18,5%‡ | 11,5% | 24,8%‡ |
| Improvement of endoscopic appearance of the mucosab | 15,6% | 31,3%† | 23,0% | 42,4%* |
| Normalisation of endoscopic appearance of the mucosac | 1,6% | 6,7%‡ | 2,5% | 10,9%‡ |
| Clinical responsed | 32,8% | 59,9%* | 34,4% | 60,7%* |
| OCTAVE induction study 2 | ||||
| Central endoscopy read | Local endoscopy read | |||
| Endpoint | Placebo | Tofacitinib 10 mg twice daily | Placebo | Tofacitinib 10 mg twice daily |
| N=112 | N=429 | N=112 | N=429 | |
| Remissiona | 3,6% | 16,6%† | 5,4% | 20,7%† |
| Improvement of endoscopic appearance of the mucosab | 11,6% | 28,4%† | 15,2% | 36,4%* |
| Normalisation of endoscopic appearance of the mucosac | 1,8% | 7,0%‡ | 0,0% | 9,1%‡ |
| Clinical responsed | 28,6% | 55,0%* | 29,5% | 58,0%* |
* p <0.0001;
† p <0.001;
‡ p <0.05.
N=number of patients in the analysis set.
a Primary endpoint: Remission was defined as clinical remission (a Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0.
b Key secondary endpoint: Improvement of endoscopic appearance of the mucosa was defined as Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern).
c Normalisation of endoscopic appearance of the mucosa was defined as a Mayo endoscopic subscore of 0.
d Clinical response was defined as a decrease from baseline in Mayo score of ≥3 points and ≥30%, with an accompanying decrease in the subscore for rectal bleeding of ≥1 point or absolute subscore for rectal bleeding of 0 or 1.
In both subgroups of patients with or without prior TNF inhibitor failure, a greater proportion of patients treated with tofacitinib 10 mg twice daily achieved remission and improvement of endoscopic appearance of the mucosa at week 8 as compared to placebo. This treatment difference was consistent between the 2 subgroups (Table 24).
Table 24. Proportion of patients meeting primary and key secondary efficacy endpoints at week 8 by TNF inhibitor therapy subgroups (OCTAVE induction study 1 and OCTAVE induction study 2, central endoscopy read):
| OCTAVE induction study 1 | |||||
|---|---|---|---|---|---|
| Endpoint | Placebo N=122 | Tofacitinib 10 mg twice daily N=476 | |||
| Remissiona | |||||
| With prior TNF inhibitor failure | 1,6% (1/64) | 11,1% (27/243) | Without prior TNF inhibitor failureb | 15,5% (9/58) | 26,2% (61/233) |
| Improvement of endoscopic appearance of the mucosac | |||||
| With prior TNF inhibitor failure | 6,3% (4/64) | 22,6% (55/243) | |||
| Without prior TNF inhibitor failureb | 25,9% (15/58) | 40,3% (94/233) | |||
| OCTAVE induction study 2 | |||||
| Endpoint | Placebo N=112 | Tofacitinib 10 mg twice daily N=429 | |||
| Remissiona | |||||
| With prior TNF inhibitor failure | 0,0% (0/60) | 11,7% (26/222) | Without prior TNF inhibitor failureb | 7,7% (4/52) | 21,7% (45/207) |
| Improvement of endoscopic appearance of the mucosac | |||||
| With prior TNF inhibitor failure | 6,7% (4/60) | 21,6% (48/222) | |||
| Without prior TNF inhibitor failureb | 17,3% (9/52) | 35,7% (74/207) | |||
TNF=tumour necrosis factor; N=number of patients in the analysis set.
a Remission was defined as clinical remission (a Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0.
b Included T NF Inhibitor naïve patients
c Improvement of endoscopic appearance of the mucosa was defined as Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern).
As early as week 2, the earliest scheduled study visit, and at each visit thereafter, significant differences were observed between tofacitinib 10 mg twice daily and placebo in the change from baseline in rectal bleeding and stool frequency, and partial Mayo score.
Maintenance (OCTAVE Sustain)
Patients who completed 8 weeks in 1 of the induction studies and achieved clinical response were re-randomised into OCTAVE Sustain; 179 out of 593 (30.2%) patients were in remission at baseline of OCTAVE Sustain.
The primary endpoint in OCTAVE Sustain was the proportion of patients in remission at week 52. The 2 key secondary endpoints were the proportion of patients with improvement of endoscopic appearance at week 52, and the proportion of patients with sustained corticosteroid-free remission at both week 24 and week 52 among patients in remission at baseline of OCTAVE Sustain.
A significantly greater proportion of patients in both the tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily treatment groups achieved the following endpoints at week 52 as compared to placebo: remission, improvement of endoscopic appearance of the mucosa, normalisation of endoscopic appearance of the mucosa, maintenance of clinical response, remission among patients in remission at baseline, and sustained corticosteroid-free remission at both week 24 and week 52 among patients in remission at baseline, as shown in Table 25.
Table 25. Proportion of patients meeting efficacy endpoints at week 52 (OCTAVE sustain):
| Central endoscopy read | Local endoscopy read | |||||
|---|---|---|---|---|---|---|
| Endpoint | Placebo N=198 | Tofacitinib 5 mg twice daily N=198 | Tofacitinib 10 mg twice daily N=197 | Placebo N=198 | Tofacitinib 5 mg twice daily N=198 | Tofacitinib 10 mg twice daily N=197 |
| Remissiona | 11,1% | 34,3%* | 40,6%* | 13,1% | 39,4%* | 47,7%* |
| Improvement of endoscopic appearance of the mucosab | 13,1% | 37,4%* | 45,7%* | 15,7% | 44,9%* | 53,8%* |
| Normalisation of endoscopic appearance of the mucosac | 4,0% | 14,6%** | 16,8%* | 5,6% | 22,2%* | 29,4%* |
| Maintenance of clinical responsed | 20,2% | 51,5%* | 61,9%* | 20,7% | 51,0%* | 61,4%* |
| Remission among patients in remission at baselinea,f | 10,2% | 46,2%* | 56,4%* | 11,9% | 50,8%* | 65,5%* |
| Sustained corticosteroid-free remission at both week 24 and week 52 among patients in remission at baseline e,f | 5,1% | 35,4%* | 47,3%* | 11,9% | 47,7%* | 58,2%* |
| Corticosteroid-free remission among patients taking corticosteroids at baselinea,g | 10,9% | 27,7%† | 27,6%† | 13,9% | 32,7%† | 31,0%† |
* p <0.0001;
** p <0.001;
† p <0.05 for tofacitinib versus placebo.
N=number of patients in the analysis set.
a Remission was defined as clinical remission (a Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0.
b Improvement of endoscopic appearance of the mucosa was defined as Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern).
c Normalisation of endoscopic appearance of the mucosa was defined as a Mayo endoscopic subscore of 0.
d Maintenance of clinical response was defined by a decrease from the induction study (OCT AVE Induction 1, OCT AVE Induction 2) baseline Mayo score of ≥3 points and ≥30%, with an accompanying decrease in the rectal bleeding subscore of ≥1 point or rectal bleeding subscore of 0 or 1. Pat ients were to be in clinical response at baseline of the maintenance study OCT AVE Sustain.
e Sustained corticosteroid-free remission was defined as being in remission and not taking corticosteroids for at least 4 weeks prior to the visit at both week 24 and week 52.
f N=59 for placebo, N=65 for tofacitinib 5 mg twice daily, N=55 for tofacitinib 10 mg twice daily.
g N=101 for placebo, N=101 for tofacitinib 5 mg twice daily, N=87 for tofacitinib 10 mg twice daily.
In both subgroups of patients with or without prior TNF inhibitor failure, a greater proportion of patients treated with either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily achieved the following endpoints at week 52 of OCTAVE Sustain as compared to placebo: remission, improvement of endoscopic appearance of the mucosa, or sustained corticosteroid-free remission at both week 24 and week 52 among patients in remission at baseline (Table 26). This treatment difference from placebo was similar between tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily in the subgroup of patients without prior TNF inhibitor failure. In the subgroup of patients with prior TNF inhibitor failure, the observed treatment difference from placebo was numerically greater for tofacitinib 10 mg twice daily than tofacitinib 5 mg twice daily by 9.7 to 16.7 percentage points across the primary and key secondary endpoints.
Table 26. Proportion of patients meeting primary and key secondary efficacy endpoints at week 52 by TNF inhibitor therapy subgroup (OCTAVE sustain, central endoscopy read):
| Endpoint | Placebo N=198 | Tofacitinib 5 mg twice daily N=198 | Tofacitinib 10 mg twice daily N=197 |
|---|---|---|---|
| Remissiona | |||
| With prior TNF inhibitor failure | 10/89 (11,2%) | 20/83 (24,1%) | 34/93 (36,6%) |
| Without prior TNF inhibitor failureb | 12/109 (11,0%) | 48/115 (41,7%) | 46/104 (44,2%) |
| Improvement of endoscopic appearance of the mucosac | |||
| With prior TNF inhibitor failure | 11/89 (12,4%) | 25/83 (30,1%) | 37/93 (39,8%) |
| Without prior TNF inhibitor failureb | 15/109 (13,8%) | 49/115 (42,6%) | 53/104 (51,0%) |
| Sustained corticosteroid-free remission at both week 24 and week 52 among patients in remission at baselined | |||
| With prior TNF inhibitor failure | 1/21 (4,8%) | 4/18 (22,2%) | 7/18 (38,9%) |
| Without prior TNF inhibitor failureb | 2/38 (5,3%) | 19/47 (40,4%) | 19/37 (51,4%) |
TNF=tumour necrosis factor; N=number of patients in the analy sis set.
a Remission was defined as clinical remission (a Mayo score ≤2 with no individual subscore >1) and rectal bleeding subscore of 0.
b Included T NF Inhibitor naïve patients.
c Improvement of endoscopic appearance of the mucosa was defined as Mayo endoscopy subscore of 0 (normal or inactive disease) or 1 (erythema, decreased vascular pattern).
d Sustained corticosteroid-free remission was defined as being in remission and not taking corticosteroids for at least 4 weeks prior to the visit at both week 24 and week 52.
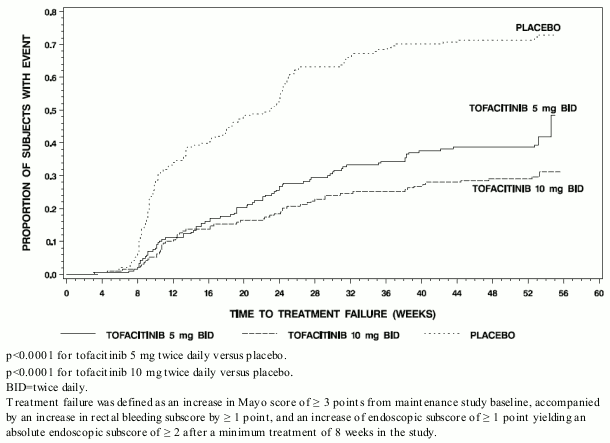
The proportion of patients in both tofacitinib groups who had treatment failure was lower compared to placebo at each time point as early as week 8, the first time point where treatment failure was assessed, as shown in Figure 2.
Figure 2. Time to treatment failure in maintenance study OCTAVE sustain (Kaplan-Meier Curves):
Health-related and quality of life outcomes
Tofacitinib 10 mg twice daily demonstrated greater improvement from baseline compared to placebo in physical component summary (PCS) and mental component summary (MCS) scores, and in all 8 domains of the SF-36 in the induction studies (OCTAVE Induction 1, OCTAVE Induction 2). In the maintenance study (OCTAVE Sustain), tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily demonstrated greater maintenance of improvement compared to placebo in PCS and MCS scores, and in all 8 domains of the SF-36 at week 24 and week 52.
Tofacitinib 10 mg twice daily demonstrated greater improvement from baseline compared to placebo at week 8 in the total and all 4 domain scores of the Inflammatory Bowel Disease Questionnaire (IBDQ) (bowel symptoms, systemic function, emotional function, and social function) in the induction studies (OCTAVE Induction 1, OCTAVE Induction 2). In the maintenance study (OCTAVE Sustain), tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily demonstrated greater maintenance of improvement compared to placebo in the total and all 4 domain scores of the IBDQ at week 24 and week 52.
Improvements were also observed in the EuroQoL 5-Dimension (EQ-5D) and various domains of the Work Productivity and Activity Impairment (WPAI-UC) questionnaire in both induction and maintenance studies compared to placebo.
Open-label extension study (OCTAVE Open)
Patients who did not achieve clinical response in one of the induction studies (OCTAVE Induction 1 or OCTAVE Induction 2) after 8 weeks of tofacitinib 10 mg twice daily were allowed to enter an open-label extension study (OCTAVE Open). After an additional 8 weeks of tofacitinib 10 mg twice daily in OCTAVE Open, 53% (154/293) patients achieved clinical response and 14% (42/293) patients achieved remission.
Patients who achieved clinical response in 1 of the induction studies (OCTAVE Induction 1 or OCTAVE Induction 2) with tofacitinib 10 mg twice daily but experienced treatment failure after their dose was reduced to tofacitinib 5 mg twice daily or following treatment interruption in OCTAVE Sustain (i.e., were randomised to placebo), had their dose increased to tofacitinib 10 mg twice daily in OCTAVE Open. After 8 weeks on tofacitinib 10 mg twice daily in OCTAVE Open, remission was achieved in 35% (20/58) patients who received tofacitinib 5 mg twice daily in OCTAVE Sustain and 40% (40/99) patients with dose interruption in OCTAVE Sustain. At month 12 in OCTAVE Open, 52% (25/48) and 45% (37/83) of these patients achieved remission, respectively.
Furthermore, at month 12 of study OCTAVE Open, 74% (48/65) of patients who achieved remission at the end of study OCTAVE Sustain on either tofacitinib 5 mg twice daily or tofacitinib 10 mg twice daily remained in remission while receiving tofacitinib 5 mg twice daily.
Paediatric population
The European Medicines Agency has deferred the obligation to submit results of studies with tofacitinib in one or more subsets of the paediatric population in other rarer types of juvenile idiopathic arthritis and in ulcerative colitis (see section 4.2 for information on paediatric use).
Polyarticular juvenile idiopathic arthritis and juvenile PsA
The tofacitinib Phase 3 program for JIA consisted of one completed Phase 3 trial (Study JIA-I [A3921104]) and one ongoing long-term extension (LTE) (A3921145) trial. In these studies the following JIA subgroups were included: patients with either RF+ or RF- polyarthritis, extended oligoarthritis, systemic JIA with active arthritis and no current systemic symptoms (referred as pJIA dataset) and two separate subgroups of patients with juvenile PsA and enthesitis-related arthritis (ERA). However, the pJIA efficacy population only includes the subgroups with either RF+ or RFpolyarthritis or extended oligoarthritis; inconclusive results have been seen in the subgroup of patients with systemic JIA with active arthritis and no current systemic symptoms. Patients with juvenile PsA are included as separate efficacy subgroup. ERA patients are not included in the efficacy analysis.
All eligible patients in Study JIA-I received open-label tofacitinib 5 mg film-coated tablets twice daily or tofacitinib oral solution weight-based equivalent twice daily for 18 weeks (run-in phase); patients who achieved at least a JIA ACR30 response at the end of the open-label phase were randomised (1:1) to either active tofacitinib 5 mg film-coated tablets or tofacitinib oral solution, or placebo in the 26-week double-blind, placebo-controlled phase. Patients who did not achieve a JIA ACR30 response at the end of the open-label run-in phase or experienced a single episode of disease flare at any time were discontinued from the study. A total of 225 patients were enrolled in the open-label run-in phase. Of these, 173 (76.9%) patients were eligible to be randomised into the double-blind phase to either active tofacitinib 5 mg film-coated tablets or tofacitinib oral solution weight-based equivalent twice daily (n=88) or placebo (n=85). There were 58 (65.9%) patients in the tofacitinib group and 58 (68.2%) patients in the placebo group taking MTX during the double-blind phase, which was permitted but not required per the protocol.
There were 133 patients with pJIA [RF+ or RF- polyarthritis and extended oligoarthritis] and 15 with juvenile PsA randomised into the double-blind phase of the study and included in the efficacy analyses presented below.
Signs and symptoms
A significantly smaller proportion of patients with pJIA in Study JIA-I treated with tofacitinib 5 mg film-coated tablets twice daily or tofacitinib oral solution weight-based equivalent twice daily flared at Week 44 compared with patients treated with placebo. A significantly greater proportion of patients with pJIA treated with tofacitinib 5 mg film-coated tablets or tofacitinib oral solution achieved JIA ACR30, 50, and 70 responses compared to patients treated with placebo at Week 44 (Table 27).
The occurrence of disease flare and JIA ACR30/50/70 results were favourable to tofacitinib 5 mg twice daily in comparison to placebo across the RF+ polyarthritis, RF- polyarthritis, extended oligoarthritis, and jPsA JIA subtypes and were consistent with those for the overall study population. The occurrence of disease flare and JIA ACR30/50/70 results were favourable to tofacitinib 5 mg twice daily in comparison to placebo for pJIA patients who received tofacitinib 5 mg twice daily with concomitant MTX use on Day 1 [n=101 (76%)] and those who were on tofacitinib monotherapy [n=32 (24%)]. In addition, the occurrence of disease flare and JIA ACR30/50/70 results were also favourable to tofacitinib 5 mg twice daily compared to placebo for pJIA patients who had prior bDMARD experience [n=39 (29%)] and those who were bDMARD naïve [n=94 (71%)].
In Study JIA-I at Week 2 of the open-label run-in phase, the JIA ACR30 response in patients with pJIA was 45.03%.
Table 27. Primary and secondary efficacy endpoints in patients with pJIA at Week 44* in Study JIA-I (all p-values <0.05):
| Primary endpoint (Type I error controlled) | Treatment group | Occurrence rate | Difference (%) from placebo (95% CI) |
|---|---|---|---|
| Occurrence of disease flare | Tofacitinib 5 mg Twice Daily (N=67) | 28% | -24.7 (-40.8, -8.5) |
| Placebo (N=66) | 53% | ||
| Secondary endpoints (Type I error controlled) | Treatment group | Response rate | Difference (%) from placebo (95% CI) |
| JIA ACR30 | Tofacitinib 5 mg Twice Daily (N=67) | 72% | 24,7 (8,50, 40,8) |
| Placebo (N=66) | 47% | ||
| JIA ACR50 | Tofacitinib 5 mg Twice Daily (N=67) | 67% | 20.2 (3.72, 36.7) |
| Placebo (N=66) | 47% | ||
| JIA ACR70 | Tofacitinib 5 mg Twice Daily (N=67) | 55% | 17.4 (0.65, 34.0) |
| Placebo (N=66) | 38% | ||
| Secondary endpoint (Type I error controlled) | Treatment group | LS mean (SEM) | Difference from placebo (95% CI) |
| Change from Double-Blind Baseline in CHAQ Disability Index | Tofacitinib 5 mg Twice Daily (N=67, n=46) | -0.11 (0.04) | -0.11 (-0.22, -0.01) |
| Placebo (N=66, n=31) | 0.00 (0.04) |
ACR = American College of Rheumatology; CHAQ = childhood health assessment questionnaire; CI = confidence interval; LS = least squares; n = number of patients with observations at the visit; N = total number of patients; JIA = juvenile idiopathic arthritis; SEM = standard error of the mean
* The 26-week double-blind phase is from Week 18 through Week 44 on and after randomisation day.
The Type-I error-controlled endpoints are tested in this order: Disease Flare, JIA ACR50, JIA ACR30, JIA ACR70, CHAQ Disability Index.
In the double-blind phase, each of the components of the JIA ACR response showed greater improvement from the open-label baseline (Day 1) at Week 24, and Week 44 for patients with pJIA treated with tofacitinib oral solution dosed as 5 mg twice daily or weight-based equivalent twice daily compared with those receiving placebo in Study JIA-I.
Physical function and health-related quality of life
Changes in physical function in Study JIA-I were measured by the CHAQ Disability Index. The mean change from the double-blind baseline in CHAQ-Disability Index in patients with pJIA was significantly lower in the tofacitinib 5 mg film-coated tablets twice daily or tofacitinib oral solution weight-based equivalent twice daily compared to placebo at Week 44 (Table 27). The mean change from the double-blind baseline in CHAQ Disability Index results were favourable to tofacitinib 5 mg twice daily in comparison to placebo across the RF+ polyarthritis, RF- polyarthritis, extended oligoarthritis, and jPsA JIA subtypes and were consistent with those for the overall study population.
Pharmacokinetic properties
The PK profile of tofacitinib is characterised by rapid absorption (peak plasma concentrations are reached within 0.5-1 hour), rapid elimination (half-life of ~3 hours) and dose-proportional increases in systemic exposure. Steady state concentrations are achieved in 24-48 hours with negligible accumulation after twice daily administration.
Absorption and distribution
Tofacitinib is well-absorbed, with an oral bioavailability of 74%. Coadministration of tofacitinib with a high-fat meal resulted in no changes in AUC while Cmax was reduced by 32%. In clinical trials, tofacitinib was administered without regard to meal.
After intravenous administration, the volume of distribution is 87 L. Approximately 40% of circulating tofacitinib is bound to plasma proteins. Tofacitinib binds predominantly to albumin and does not appear to bind to α1-acid glycoprotein. Tofacitinib distributes equally between red blood cells and plasma.
Biotransformation and elimination
Clearance mechanisms for tofacitinib are approximately 70% hepatic metabolism and 30% renal excretion of the parent drug. The metabolism of tofacitinib is primarily mediated by CYP3A4 with minor contribution from CYP2C19. In a human radiolabelled study, more than 65% of the total circulating radioactivity was accounted for by unchanged active substance, with the remaining 35% attributed to 8 metabolites, each accounting for less than 8% of total radioactivity. All metabolites have been observed in animal species and are predicted to have less than 10-fold potency than tofacitinib for JAK1/3 inhibition. No evidence of stereo conversion in human samples was detected. The pharmacologic activity of tofacitinib is attributed to the parent molecule. In vitro, tofacitinib is a substrate for MDR1, but not for breast cancer resistance protein (BCRP), OATP1B1/1B3, or OCT1/2.
Pharmacokinetics in patients
The enzymatic activity of CYP enzymes is reduced in RA patients due to chronic inflammation. In RA patients, the oral clearance of tofacitinib does not vary with time, indicating that treatment with tofacitinib does not normalise CYP enzyme activity.
Population PK analysis in RA patients indicated that systemic exposure (AUC) of tofacitinib in the extremes of body weight (40 kg, 140 kg) were similar (within 5%) to that of a 70 kg patient. Elderly patients 80 years of age were estimated to have less than 5% higher AUC relative to the mean age of 55 years. Women were estimated to have 7% lower AUC compared to men. The available data have also shown that there are no major differences in tofacitinib AUC between White, Black and Asian patients. An approximate linear relationship betw een body weight and volume of distribution was observed, resulting in higher peak (Cmax) and lower trough (Cmin) concentrations in lighter patients. However, this difference is not considered to be clinically relevant. The between-subject variability (percentage coefficient of variation) in AUC of tofacitinib is estimated to be approximately 27%.
Results from population PK analysis in patients with active PsA or moderate to severe UC were consistent with those in patients with RA.
Renal impairment
Subjects with mild (creatinine clearance 50-80 mL/min), moderate (creatinine clearance 30-49 mL/min), and severe (creatinine clearance <30 mL/min) renal impairment had 37%, 43% and 123% higher AUC, respectively, compared to subjects with normal renal function (see section 4.2). In subjects with end-stage renal disease (ESRD), contribution of dialysis to the total clearance of tofacitinib was relatively small. Following a single dose of 10 mg, mean AUC in subjects with ESRD based on concentrations measured on a non-dialysis day was approximately 40% (90% confidence intervals: 1.5-95%) higher compared to subjects with normal renal function. In clinical trials, tofacitinib was not evaluated in patients with baseline creatinine clearance values (estimated by Cockroft-Gault equation) less than 40 mL/min (see section 4.2).
Hepatic impairment
Subjects with mild (Child Pugh A) and moderate (Child Pugh B) hepatic impairment had 3%, and 65% higher AUC, respectively, compared to subjects with normal hepatic function. In clinical trials, tofacitinib was not evaluated in subjects with severe (Child Pugh C) hepatic impairment (see sections 4.2 and 4.4), or in patients screened positive for hepatitis B or C.
interactions
Tofacitinib is not an inhibitor or inducer of CYPs (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) and is not an inhibitor of UGTs (UGT1A1, UGT1A4, UGT1A6, UGT1A9, and UGT2B7). Tofacitinib is not an inhibitor of MDR1, OATP1B1/1B3, OCT2, OAT1/3, or MRP at clinically meaningful concentrations.
Comparison of PK of prolonged-release and film-coated tablet formulations
Tofacitinib 11 mg prolonged-release tablets once daily have demonstrated PK equivalence (AUC and Cmax) to tofacitinib 5 mg film-coated tablets twice daily.
Paediatric population
Pharmacokinetics in paediatric patients with juvenile idiopathic arthritis
Population PK analysis based on results from both tofacitinib 5 mg film-coated tablets twice daily and tofacitinib oral solution weight-based equivalent twice daily indicated that tofacitinib clearance and volume of distribution both decreased with decreasing body weight in JIA patients. The available data indicated that there were no clinically relevant differences in tofacitinib exposure (AUC), based on age, race, gender, patient type or baseline disease severity. The between-subject variability (% coefficient of variation) in (AUC) was estimated to be approximately 24%.
Preclinical safety data
In non-clinical studies, effects were observed on the immune and haematopoietic systems that were attributed to the pharmacological properties (JAK inhibition) of tofacitinib. Secondary effects from immunosuppression, such as bacterial and viral infections and lymphoma were observed at clinically relevant doses. Lymphoma was observed in 3 of 8 adult monkeys at 6 or 3 times the clinical tofacitinib exposure level (unbound AUC in humans at a dose of 5 mg or 10 mg twice daily), and 0 of 14 juvenile monkeys at 5 or 2.5 times the clinical exposure level of 5 mg or 10 mg twice daily. Exposure in monkeys at the no observed adverse effect level (NOAEL) for the lymphomas was approximately 1 or 0.5 times the clinical exposure level of 5 mg or 10 mg twice daily. Other findings at doses exceeding human exposures included effects on the hepatic and gastrointestinal systems.
Tofacitinib is not mutagenic or genotoxic based on the results of a series of in vitro and in vivo tests for gene mutations and chromosomal aberrations.
The carcinogenic potential of tofacitinib was assessed in 6-month rasH2 transgenic mouse carcinogenicity and 2-year rat carcinogenicity studies. Tofacitinib was not carcinogenic in mice at exposures up to 38 or 19 times the clinical exposure level at 5 mg or 10 mg twice daily. Benign testicular interstitial (Leydig) cell tumours were observed in rats: benign Leydig cell tumours in rats are not associated with a risk of Leydig cell tumours in humans. Hibernomas (malignancy of brown adipose tissue) were observed in female rats at exposures greater than or equal to 83 or 41 times the clinical exposure level at 5 mg or 10 mg twice daily. Benign thymomas were observed in female rats at 187 or 94 times the clinical exposure level at 5 mg or 10 mg twice daily.
Tofacitinib was shown to be teratogenic in rats and rabbits, and have effects in rats on female fertility (decreased pregnancy rate; decreases in the numbers of corpora lutea, implantation sites, and viable foetuses; and an increase in early resorptions), parturition, and peri/postnatal development. Tofacitinib had no effects on male fertility, sperm motility or sperm concentration. Tofacitinib was secreted in milk of lactating rats at concentrations approximately 2-fold those in serum from 1 to 8 hours postdose.
No tofacitinib-related findings were observed in juvenile animal studies that indicate a higher sensitivity of paediatric populations compared with adults. In the juvenile rat fertility study, there was no evidence of developmental toxicity, no effects on sexual maturation, and no evidence of reproductive toxicity (mating and fertility) was noted after sexual maturity. In 1-month juvenile rat and 39-week juvenile monkey studies tofacitinib-related effects on immune and haematology parameters consistent with JAK1/3 and JAK2 inhibition were observed. These effects were reversible and consistent with those also observed in adult animals at similar exposures.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.