XELJANZ Film-coated tablet Ref.[8636] Active ingredients: Tofacitinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
Contraindications
- Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
- Active tuberculosis (TB), serious infections such as sepsis, or opportunistic infections (see section 4.4).
- Severe hepatic impairment (see section 4.2).
- Pregnancy and lactation (see section 4.6).
Special warnings and precautions for use
Tofacitinib should only be used if no suitable treatment alternatives are available in patients:
- 65 years of age and older;
- patients with history of atherosclerotic cardiovascular disease or other cardiovascular risk factors (such as current or past long-time smokers);
- patients with malignancy risk factors (e.g. current malignancy or history of malignancy)
Use in patients 65 years of age and older
Considering the increased risk of serious infections, myocardial infarction, malignancies and all cause mortality with tofacitinib in patients 65 years of age and older, tofacitinib should only be used in these patients if no suitable treatment alternatives are available (see further details below in section 4.4 and section 5.1).
Combination with other therapies
Tofacitinib has not been studied and its use should be avoided in combination with biologics such as TNF antagonists, interleukin (IL)-1R antagonists, IL-6R antagonists, anti-CD20 monoclonal antibodies, IL-17 antagonists, IL-12/IL-23 antagonists, anti-integrins, selective co-stimulation modulators and potent immunosuppressants such as azathioprine, 6-mercaptopurine, ciclosporine and tacrolimus because of the possibility of increased immunosuppression and increased risk of infection.
There was a higher incidence of adverse events for the combination of tofacitinib with MTX versus tofacitinib as monotherapy in RA clinical studies.
The use of tofacitinib in combination with phosphodiesterase 4 inhibitors has not been studied in tofacitinib clinical studies.
Venous thromboembolism (VTE)
Serious VTE events including pulmonary embolism (PE), some of which were fatal, and deep vein thrombosis (DVT), have been observed in patients taking tofacitinib. In a randomised post-authorisation safety study in patients with rheumatoid arthritis who were 50 years of age or older with at least one additional cardiovascular risk factor, a dose dependent increased risk for VTE was observed with tofacitinib compared to TNF inhibitors (see sections 4.8 and 5.1).
In a post hoc exploratory analysis within this study, in patients with known VTE risk factors, occurrences of subsequent VTEs were observed more frequently in tofacitinib-treated patients that, at 12 months treatment, had D-dimer level ≥2× ULN versus those with D-dimer level <2× ULN; this was not evident in TNF inhibitor-treated patients. Interpretation is limited by the low number of VTE events and restricted D-dimer test availability (only assessed at Baseline, Month 12, and at the end of the study). In patients who did not have a VTE during the study, mean D-dimer levels were significantly reduced at Month 12 relative to Baseline across all treatment arms. However, D-dimer levels ≥2× ULN at Month 12 were observed in approximately 30% of patients without subsequent VTE events, indicating limited specificity of D-Dimer testing in this study.
Tofacitinib 10 mg twice daily for maintenance treatment is not recommended in patients with UC who have known VTE, MACE and malignancy risk factors, unless there is no suitable alternative treatment available (see section 4.2).
In patients with cardiovascular or malignancy risk factors (see also section 4.4 "Major adverse cardiovascular events (MACE)" and "Malignancy") tofacitinib should only be used if no suitable treatment alternatives are available.
In patients with VTE risk factors other than MACE or malignancy risk factors, tofacitinib should be used with caution. VTE risk factors other than MACE or malignancy risk factors include previous VTE, patients undergoing major surgery, immobilisation, use of combined hormonal contraceptives or hormone replacement therapy, inherited coagulation disorder. Patients should be re-evaluated periodically during tofacitinib treatment to assess for changes in VTE risk.
For patients with RA with known risk factors for VTE, consider testing D-dimer levels after approximately 12 months of treatment. If D-dimer test result is ≥ 2× ULN, confirm that clinical benefits outweigh risks prior to a decision on treatment continuation with tofacitinib.
Promptly evaluate patients with signs and symptoms of VTE and discontinue tofacitinib in patients with suspected VTE, regardless of dose or indication.
Retinal venous thrombosis
Retinal venous thrombosis (RVT) has been reported in patients treated with tofacitinib (see section 4.8). The patients should be advised to promptly seek medical care in case they experience symptoms suggestive of RVT.
Serious infections
Serious and sometimes fatal infections due to bacterial, mycobacterial, invasive fungal, viral, or other opportunistic pathogens have been reported in patients receiving tofacitinib (see section 4.8). The risk of opportunistic infections is higher in Asian geographic regions (see section 4.8). Rheumatoid arthritis patients taking corticosteroids may be predisposed to infection.
Tofacitinib should not be initiated in patients with active infections, including localised infections.
The risks and benefits of treatment should be considered prior to initiating tofacitinib in patients:
- with recurrent infections,
- with a history of a serious or an opportunistic infection,
- who have resided or travelled in areas of endemic mycoses,
- who have underlying conditions that may predispose them to infection.
Patients should be closely monitored for the development of signs and symptoms of infection during and after treatment with tofacitinib. Treatment should be interrupted if a patient develops a serious infection, an opportunistic infection, or sepsis. A patient who develops a new infection during treatment with tofacitinib should undergo prompt and complete diagnostic testing appropriate for an immunocompromised patient, appropriate antimicrobial therapy should be initiated, and the patient should be closely monitored.
As there is a higher incidence of infections in the elderly and in the diabetic populations in general, caution should be used when treating the elderly and patients with diabetes (see section 4.8). In patients over 65 years of age tofacitinib should only be considered if no suitable alternative treatment is available (see section 5.1).
Risk of infection may be higher with increasing degrees of lymphopenia and consideration should be given to lymphocyte counts when assessing individual patient risk of infection. Discontinuation and monitoring criteria for lymphopenia are discussed in section 4.2.
Tuberculosis
The risks and benefits of treatment should be considered prior to initiating tofacitinib in patients:
- who have been exposed to TB,
- who have resided or travelled in areas of endemic TB.
Patients should be evaluated and tested for latent or active infection prior to and per applicable guidelines during administration of tofacitinib.
Patients with latent TB, who test positive, should be treated with standard antimycobacterial therapy before administering tofacitinib.
Antituberculosis therapy should also be considered prior to administration of tofacitinib in patients who test negative for TB but who have a past history of latent or active TB and where an adequate course of treatment cannot be confirmed; or those who test negative but who have risk factors for TB infection. Consultation with a healthcare professional with expertise in the treatment of TB is recommended to aid in the decision about whether initiating antituberculosis therapy is appropriate for an individual patient. Patients should be closely monitored for the development of signs and symptoms of TB, including patients who tested negative for latent TB infection prior to initiating therapy.
Viral reactivation
Viral reactivation and cases of herpes virus reactivation (e.g., herpes zoster) were observed in clinical studies with tofacitinib. In patients treated with tofacitinib, the incidence of herpes zoster appears to be increased in:
- Japanese or Korean patients.
- Patients with an ALC less than 1,000 cells/mm³ (see section 4.2).
- Patients with long standing RA who have previously received two or more biological disease modifying antirheumatic drugs (DMARDs).
- Patients treated with 10 mg twice daily.
The impact of tofacitinib on chronic viral hepatitis reactivation is unknown. Patients screened positive for hepatitis B or C were excluded from clinical trials. Screening for viral hepatitis should be performed in accordance with clinical guidelines before starting therapy with tofacitinib.
Major adverse cardiovascular events (including myocardial infarction)
Major adverse cardiovascular events (MACE) have been observed in patients taking tofacitinib.
In a randomised post authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, an increased incidence of myocardial infarctions was observed with tofacitinib compared to TNF inhibitors (see sections 4.8 and 5.1). In patients 65 years of age and older, patients who are current or past long-time smokers, and patients with history of atherosclerotic cardiovascular disease or other cardiovascular risk factors, tofacitinib should only be used if no suitable treatment alternatives are available (see section 5.1).
Malignancy and lymphoproliferative disorder
Tofacitinib may affect host defences against malignancies.
In a randomised post authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, an increased incidence of malignancies, particularly NMSC, lung cancer and lymphoma, was observed with tofacitinib compared to TNF inhibitors (see sections 4.8 and 5.1).
NMSC lung cancers and lymphoma in patients treated with tofacitinib have also been observed in other clinical studies and in the post marketing setting.
Other malignancies in patients treated with tofacitinib were observed in clinical studies and the post-marketing setting, including, but not limited to, breast cancer, melanoma, prostate cancer, and pancreatic cancer.
In patients 65 years of age and older, patients who are current or past long-time smokers, and patients with other malignancy risk factors (e.g. current malignancy or history of malignancy other than a successfully treated non-melanoma skin cancer) tofacitinib should only be used if no suitable treatment alternatives are available (see section 5.1). Periodic skin examination is recommended for all patients, particularly those who are at increased risk for skin cancer (see Table 8 in section 4.8).
Interstitial lung disease
Caution is also recommended in patients with a history of chronic lung disease as they may be more prone to infections. Events of interstitial lung disease (some of which had a fatal outcome) have been reported in patients treated with tofacitinib in RA clinical trials and in the post-marketing setting although the role of Janus kinase (JAK) inhibition in these events is not known. Asian RA patients are known to be at higher risk of interstitial lung disease, thus caution should be exercised in treating these patients.
Gastrointestinal perforations
Events of gastrointestinal perforation have been reported in clinical trials although the role of JAK inhibition in these events is not known. Tofacitinib should be used with caution in patients who may be at increased risk for gastrointestinal perforation (e.g. patients with a history of diverticulitis, patients with concomitant use of corticosteroids and/or nonsteroidal anti-inflammatory drugs). Patients presenting with new onset abdominal signs and symptoms should be evaluated promptly for early identification of gastrointestinal perforation.
Fractures
Fractures have been observed in patients treated with tofacitinib.
Tofacitinib should be used with caution in patients with known risk factors for fractures such as elderly patients, female patients and patients with corticosteroid use, regardless of indication and dosage.
Liver enzymes
Treatment with tofacitinib was associated with an increased incidence of liver enzyme elevation in some patients (see section 4.8 liver enzyme tests). Caution should be exercised when considering initiation of tofacitinib treatment in patients with elevated alanine aminotransferase (ALT) or aspartate aminotransferase (AST), particularly when initiated in combination with potentially hepatotoxic medicinal products such as MTX. Following initiation, routine monitoring of liver tests and prompt investigation of the causes of any observed liver enzyme elevations are recommended to identify potential cases of drug-induced liver injury. If drug-induced liver injury is suspected, the administration of tofacitinib should be interrupted until this diagnosis has been excluded.
Hypersensitivity
In post-marketing experience, cases of drug hypersensitivity associated with tofacitinib administration have been reported. Allergic reactions included angioedema and urticaria; serious reactions have occurred. If any serious allergic or anaphylactic reaction occurs, tofacitinib should be discontinued immediately.
Laboratory parameters
Lymphocytes
Treatment with tofacitinib was associated with an increased incidence of lymphopenia compared to placebo. Lymphocyte counts less than 750 cells/mm³ were associated with an increased incidence of serious infections. It is not recommended to initiate or continue tofacitinib treatment in patients with a confirmed lymphocyte count less than 750 cells/mm³. Lymphocytes should be monitored at baseline and every 3 months thereafter. For recommended modifications based on lymphocyte counts, see section 4.2.
Neutrophils
Treatment with tofacitinib was associated with an increased incidence of neutropenia (less than 2,000 cells/mm³) compared to placebo. It is not recommended to initiate tofacitinib treatment in patients with an ANC less than 1,000 cells/mm³. ANC should be monitored at baseline and after 4 to 8 weeks of treatment and every 3 months thereafter. For recommended modifications based on ANC, see section 4.2.
Haemoglobin
Treatment with tofacitinib has been associated with decreases in haemoglobin levels. It is not recommended to initiate tofacitinib treatment in patients with a haemoglobin value less than 9 g/dL. Haemoglobin should be monitored at baseline and after 4 to 8 weeks of treatment and every 3 months thereafter. For recommended modifications based on haemoglobin level, see section 4.2.
Lipid monitoring
Treatment with tofacitinib was associated with increases in lipid parameters such as total cholesterol, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. Maximum effects were generally observed within 6 weeks. Assessment of lipid parameters should be performed after 8 weeks following initiation of tofacitinib therapy. Patients should be managed according to clinical guidelines for the management of hyperlipidaemia. Increases in total and LDL cholesterol associated with tofacitinib may be decreased to pretreatment levels with statin therapy.
Hypoglycaemia in patients treated for diabetes
There have been reports of hypoglycaemia following initiation of tofacitinib in patients receiving medication for diabetes. Dose adjustment of anti-diabetic medication may be necessary in the event that hypoglycaemia occurs.
Vaccinations
Prior to initiating tofacitinib, it is recommended that all patients be brought up to date with all immunisations in agreement with current immunisation guidelines. It is recommended that live vaccines not be given concurrently with tofacitinib. The decision to use live vaccines prior to tofacitinib treatment should take into account the pre-existing immunosuppression in a given patient.
Prophylactic zoster vaccination should be considered in accordance with vaccination guidelines. Particular consideration should be given to patients with longstanding RA who have previously received two or more biological DMARDs. If live zoster vaccine is administered; it should only be administered to patients with a known history of chickenpox or those that are seropositive for varicella zoster virus (VZV). If the history of chickenpox is considered doubtful or unreliable it is recommended to test for antibodies against VZV.
Vaccination with live vaccines should occur at least 2 weeks but preferably 4 weeks prior to initiation of tofacitinib or in accordance with current vaccination guidelines regarding immunomodulatory medicinal products. No data are available on the secondary transmission of infection by live vaccines to patients receiving tofacitinib.
Excipients contents
This medicinal product contains lactose. Patients with rare hereditary problems of galactose intolerance, total lactase deficiency or glucose-galactose malabsorption should not take this medicinal product. This medicinal product contains less than 1 mmol sodium (23 mg) per tablet, that is to say essentially 'sodium-free'.
Interaction with other medicinal products and other forms of interaction
Potential for other medicinal products to influence the pharmacokinetics (PK) of tfacitinib
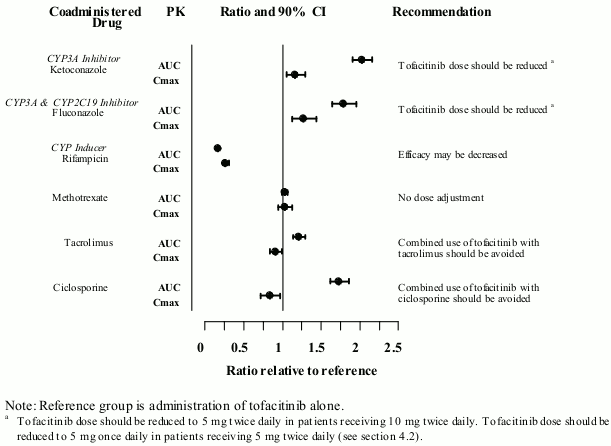
Since tofacitinib is metabolised by CYP3A4, interaction with medicinal products that inhibit or induce CYP3A4 is likely. Tofacitinib exposure is increased when coadministered with potent inhibitors of CYP3A4 (e.g. ketoconazole) or when administration of one or more concomitant medicinal products results in both moderate inhibition of CYP3A4 and potent inhibition of CYP2C19 (e.g. fluconazole) (see section 4.2).
Tofacitinib exposure is decreased when coadministered with potent CYP inducers (e.g. rifampicin). Inhibitors of CYP2C19 alone or P-glycoprotein are unlikely to significantly alter the PK of tofacitinib.
Coadministration with ketoconazole (strong CYP3A4 inhibitor), fluconazole (moderate CYP3A4 and potent CYP2C19 inhibitor), tacrolimus (mild CYP3A4 inhibitor) and c iclosporine (moderate CYP3A4 inhibitor) increased tofacitinib AUC, while rifampicin (potent CYP inducer) decreased tofacitinib AUC. Coadministration of tofacitinib with potent CYP inducers (e.g. rifampicin) may result in a loss of or reduced clinical response (see Figure 1). Coadministration of potent inducers of CYP3A4 with tofacitinib is not recommended. Coadministration with ketoconazole and fluconazole increased tofacitinib Cmax, while tacrolimus, ciclosporine and rifampicin decreased tofacitinib Cmax. Concomitant administration with MTX 15-25 mg once weekly had no effect on the PK of tofacitinib in RA patients (see Figure 1).
Figure 1. Impact of other medicinal products on PK of tofacitinib:
Potential for tofacitinib to influence the PK of other medicinal products
Coadministration of tofacitinib did not have an effect on the PK of oral contraceptives, levonorgestrel and ethinyl estradiol, in healthy female volunteers.
In RA patients, coadministration of tofacitinib with MTX 15-25 mg once weekly decreased the AUC and Cmax of MTX by 10% and 13%, respectively. The extent of decrease in MTX exposure does not warrant modifications to the individualised dosing of MTX.
Paediatric population
Interaction studies have only been performed in adults.
Fertility, pregnancy and lactation
Pregnancy
There are no adequate and well-controlled studies on the use of tofacitinib in pregnant women. Tofacitinib has been shown to be teratogenic in rats and rabbits, and to affect parturition and peri/postnatal development (see section 5.3).
As a precautionary measure, the use of tofacitinib during pregnancy is contraindicated (see section 4.3).
Women of childbearing potential/contraception in females
Women of childbearing potential should be advised to use effective contraception during treatment with tofacitinib and for at least 4 weeks after the last dose.
Breast-feeding
It is not known whether tofacitinib is secreted in human milk. A risk to the breast-fed child cannot be excluded. Tofacitinib was secreted in the milk of lactating rats (see section 5.3). As a precautionary measure, the use of tofacitinib during breast-feeding is contraindicated (see section 4.3).
Fertility
Formal studies of the potential effect on human fertility have not been conducted. Tofacitinib impaired female fertility but not male fertility in rats (see section 5.3).
Effects on ability to drive and use machines
Tofacitinib has no or negligible influence on the ability to drive and use machines.
Undesirable effects
Summary of the safety profile
Rheumatoid arthritis
The most common serious adverse reactions were serious infections (see section 4.4). In the long-term safety all exposure population, the most common serious infections reported with tofacitinib were pneumonia (1.7%), herpes zoster (0.6%), urinary tract infection (0.4%), cellulitis (0.4%), diverticulitis (0.3%), and appendicitis (0.2%). Among opportunistic infections, TB and other mycobacterial infections, cryptococcus, histoplasmosis, oesophageal candidiasis, multidermatomal herpes zoster, cytomegalovirus, BK virus infections and listeriosis were reported with tofacitinib. Some patients have presented with disseminated rather than localised disease. Other serious infections that were not reported in clinical studies may also occur (e.g., coccidioidomycosis).
The most commonly reported adverse reactions during the first 3 months in controlled clinical trials were headache (3.9%), upper respiratory tract infections (3.8%), viral upper respiratory tract infection (3.3%), diarrhoea (2.9%), nausea (2.7%), and hypertension (2.2%).
The proportion of patients who discontinued treatment due to adverse reactions during first 3 months of the double-blind, placebo or MTX controlled studies was 3.8% for patients taking tofacitinib. The most common infections resulting in discontinuation of therapy during the first 3 months in controlled clinical trials were herpes zoster (0.19%) and pneumonia (0.15%).
Psoriatic arthritis
Overall, the safety profile observed in patients with active PsA treated with tofacitinib was consistent with the safety profile observed in patients with RA treated with tofacitinib.
Ankylosing spondylitis
Overall, the safety profile observed in patients with active AS treated with tofacitinib was consistent with the safety profile observed in patients with RA treated with tofacitinib.
Ulcerative colitis
The most commonly reported adverse reactions in patients receiving tofacitinib 10 mg twice daily in the induction studies were headache, nasopharyngitis, nausea, and arthralgia.
In the induction and maintenance studies, across tofacitinib and placebo treatment groups, the most common categories of serious adverse reactions were gastrointestinal disorders and infections, and the most common serious adverse reaction was worsening of UC.
Overall, the safety profile observed in patients with UC treated with tofacitinib was consistent with the safety profile of tofacitinib in the RA indication.
Tabulated list of adverse reactions
The ADRs listed in the table below are from clinical studies in patients with RA, PsA, and UC and are presented by System Organ Class (SOC) and frequency categories, defined using the following convention: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (≥1/10,000 to <1/1,000), very rare (<1/10,000), or not known (cannot be estimated from the available data). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Table 8. Adverse drug reactions:
Infections and infestations
Common: Pneumonia, Influenza, Herpes zoster, Urinary tract infection, Sinusitis, Bronchitis, Nasopharyngitis, Pharyngitis
Uncommon: Tuberculosis, Diverticulitis, Pyelonephritis, Cellulitis, Herpes simplex, Gastroenteritis viral, Viral infection
Rare: Sepsis, Urosepsis, Disseminated TB Bacteraemia, Pneumocystis jirovecii pneumonia, Pneumonia pneumococcal, Pneumonia bacterial, Cytomegalovirus infection, Arthritis bacterial
Very rare: Tuberculosis of central nervous system, Meningitis cryptococcal, Necrotizing fasciitis, Encephalitis, Staphylococcal bacteraemia, Mycobacterium avium complex infection, Atypical mycobacterial infection
Neoplasms benign, malignant and unspecified (incl cysts and polyps)
Uncommon: Lung cancer, Non-melanoma skin cancers
Rare: Lymphoma
Blood and lymphatic system disorders
Common: Lymphopenia, Anaemia
Uncommon: Leukopenia, Neutropenia
Immune system disorders
Not known: Hypersensitivity*, Angioedema*, Urticaria*
Metabolism and nutrition disorders
Uncommon: Dyslipidaemia, Hyperlipidaemia, Dehydration
Psychiatric disorders
Uncommon: Insomnia
Nervous system disorders
Common: Headache
Uncommon: Paraesthesia
Cardiac disorders
Uncommon: Myocardial infarction
Vascular disorders
Common: Hypertension
Uncommon: Venous thromboembolism**
Respiratory, thoracic and mediastinal disorders
Common: Cough
Uncommon: Dyspnoea, Sinus congestion
Gastrointestinal disorders
Common: Abdominal pain, Vomiting, Diarrhoea, Nausea, Gastritis, Dyspepsia
Hepatobiliary disorders
Uncommon: Hepatic steatosis, Hepatic enzyme increased, Transaminases increased, Liver function test abnormal, Gamma glutamyltransferase increased
Rare: Liver function test abnormal
Skin and subcutaneous tissue disorders
Common: Rash, Acne
Uncommon: Erythema, Pruritus
Musculoskeletal and connective tissue disorders
Common: Arthralgia
Uncommon: Joint swelling, Tendonitis
Rare: Musculoskeletal pain
General disorders and administration site conditions
Common: Oedema peripheral
Uncommon: Pyrexia, Fatigue
Investigations
Common: Blood creatine phosphokinase increased
Uncommon: Blood creatinine increased, Blood cholesterol increased, Low density lipoprotein increased, Weight increased
Injury, poisoning and procedural complications
Uncommon: Ligament sprain, Muscle strain
* Spontaneous reporting data
** Venous thromboembolism includes PE and DVT
Description of selected adverse reactions
Venous thromboembolism
Rheumatoid arthritis
In a large (N=4,362), randomised post-authorisation safety study of rheumatoid arthritis patients who were 50 years of age and older and had at least one additional cardiovascular (CV) risk factor, VTE was observed at an increased and dose-dependent incidence in patients treated with tofacitinib compared to TNF inhibitors (see section 5.1). The majority of these events were serious and some resulted in death. The incidence rates (95% CI) for PE for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors were 0.17 (0.08-0.33), 0.50 (0.32-0.74), and 0.06 (0.01-0.17) patients with events per 100 patient-years, respectively. Compared with TNF inhibitors, the hazard ratio (HR) for PE was 2.93 (0.79-10.83) and 8.26 (2.49, 27.43) for tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily, respectively (see section 5.1). In tofacitinib-treated patients where PE was observed, the majority (97%) had VTE risk factors.
Ankylosing spondylitis
In the combined Phase 2 and Phase 3 randomised controlled clinical studies, there were no VTE events in 420 patients (233 patient-years of observation) receiving tofacitinib up to 48 weeks.
Ulcerative colitis (UC)
In the UC ongoing extension trial, cases of PE and DVT have been observed in patients using tofacitinib 10 mg twice daily and with underlying VTE risk factor(s).
Overall infections
Rheumatoid arthritis
In controlled phase 3 clinical studies, the rates of infections over 0-3 months in the 5 mg twice daily (total 616 patients) and 10 mg twice daily (total 642 patients) tofacitinib monotherapy groups were 16.2% (100 patients) and 17.9% (115 patients), respectively, compared to 18.9% (23 patients) in the placebo group (total 122 patients). In controlled phase 3 clinical studies with background DMARDs, the rates of infections over 0-3 months in the 5 mg twice daily (total 973 patients) and 10 mg twice daily (total 969 patients) tofacitinib plus DMARD group were 21.3% (207 patients) and 21.8% (211 patients), respectively, compared to 18.4% (103 patients) in the placebo plus DMARD group (total 559 patients).
The most commonly reported infections were upper respiratory tract infections and nasopharyngitis (3.7% and 3.2%, respectively).
The overall incidence rate of infections with tofacitinib in the long-term safety all exposure population (total 4,867 patients) was 46.1 patients with events per 100 patient-years (43.8 and 47.2 patients with events for 5 mg and 10 mg twice daily, respectively). For patients (total 1,750) on monotherapy, the rates were 48.9 and 41.9 patients with events per 100 patient-years for 5 mg and 10 mg twice daily, respectively. For patients (total 3,117) on background DMARDs, the rates were 41.0 and 50.3 patients with events per 100 patient-years for 5 mg and 10 mg twice daily, respectively.
Ankylosing spondylitis
In the combined Phase 2 and Phase 3 clinical studies, during the placebo-controlled period of up to 16 weeks, the frequency of infections in the tofacitinib 5 mg twice daily group (185 patients) was 27.6% and the frequency in the placebo group (187 patients) was 23.0%. In the combined Phase 2 and Phase 3 clinical studies, among the 316 patients treated with tofacitinib 5 mg twice daily for up to 48 weeks, the frequency of infections was 35.1%.
Ulcerative colitis
In the randomised 8-week Phase ⅔ induction studies, the proportions of patients with infections were 21.1% (198 patients) in the tofacitinib 10 mg twice daily group compared to 15.2% (43 patients) in the placebo group. In the randomised 52-week phase 3 maintenance study, the proportion of patients with infections were 35.9% (71 patients) in the 5 mg twice daily and 39.8% (78 patients) in the 10 mg twice daily tofacitinib groups, compared to 24.2% (48 patients) in the placebo group.
In the entire treatment experience with tofacitinib, the most commonly reported infection was nasopharyngitis, occurring in 18.2% of patients (211 patients).
In the entire treatment experience with tofacitinib, the overall incidence rate of infections was 60.3 events per 100 patient-years (involving 49.4% of patients; total 572 patients).
Serious infections
Rheumatoid arthritis
In the 6-month and 24-month, controlled clinical studies, the rate of serious infections in the 5 mg twice daily tofacitinib monotherapy group was 1.7 patients with events per 100 patient-years. In the 10 mg twice daily tofacitinib monotherapy group the rate was 1.6 patients with events per 100 patient-years, the rate was 0 events per 100 patient-years for the placebo group, and the rate was 1.9 patients with events per 100 patient-years for the MTX group.
In studies of 6-, 12-, or 24-month duration, the rates of serious infections in the 5 mg twice daily and 10 mg twice daily tofacitinib plus DMARD groups were 3.6 and 3.4 patients with events per 100 patient-years, respectively,
In the long-term safety all exposure population, the overall rates of serious infections were 2.4 and 3.0 patients with events per 100 patient-years for 5 mg and 10 mg twice daily tofacitinib groups, respectively. The most common serious infections included pneumonia, herpes zoster, urinary tract infection, cellulitis, gastroenteritis and diverticulitis. Cases of opportunistic infections have been reported (see section 4.4).
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years or older with at least one additional cardiovascular risk factor, a dose-dependent increase in serious infections was observed with tofacitinib compared to TNF inhibitors (see section 4.4).
The incidence rates (95% CI) for serious infections for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors were 2.86 (2.41, 3.37), 3.64 (3.11, 4.23), and 2.44 (2.02, 2.92) patients with events per 100 patient-years, respectively. Compared with TNF inhibitors, the hazard ratio (HR) for serious infections was 1.17 (0.92, 1.50) and 1.48 (1.17, 1.87) for tofacitinib 10 mg twice daily and tofacitinib 5 mg twice daily, respectively.
η4. Ankylosing spondylitis
In the combined Phase 2 and Phase 3 clinical studies, among the 316 patients treated with tofacitinib 5 mg twice daily for up to 48 weeks, there was one serious infection (aseptic meningitis) yielding a rate of 0.43 patients with events per 100 patient-years.
Ulcerative colitis
The incidence rates and types of serious infections in the UC clinical studies were generally similar to those reported in RA clinical studies with tofacitinib monotherapy treatment groups.
Serious infections in the elderly
Of the 4,271 patients who enrolled in RA studies I-VI (see section 5.1), a total of 608 RA patients were 65 years of age and older, including 85 patients 75 years and older. The frequency of serious infection among tofacitinib-treated patients 65 years of age and older was higher than those under the age of 65 (4.8 per 100 patient-years versus 2.4 per 100 patient-years, respectively).
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years or older with at least one additional cardiovascular risk factor, an increase in serious infections was observed in patients 65 years of age and older for tofacitinib 10 mg twice daily compared to TNF inhibitors and to tofacitinib 5 mg twice daily (see section 4.4). The incidence rates (95% CI) for serious infections in patients ≥65 years were 4.03 (3.02, 5.27), 5.85 (4.64, 7.30), and 3.73 (2.81, 4.85) patients with events per 100 patient-years for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors, respectively.
Compared with TNF inhibitors, the hazard ratio (HR) for serious infections in patients ≥65 years of age was 1.08 (0.74, 1.58) and 1.55 (1.10, 2.19) for tofacitinib 5 mg twice daily and tofacitinib 10 mg twice daily, respectively.
Serious infections from non-interventional post approval safety study
Data from a non-interventional post approval safety study that evaluated tofacitinib in RA patients from a registry (US Corrona) showed that a numerically higher incidence rate of serious infection was observed for the 11 mg prolonged-release tablet administered once daily than the 5 mg film-coated tablet administered twice daily. Crude incidence rates (95% CI) (i.e., not adjusted for age or sex) from availability of each formulation at 12 months following initiation of treatment were 3.45 (1.93, 5.69) and 2.78 (1.74, 4.21) and at 36 months were 4.71 (3.08, 6.91) and 2.79 (2.01, 3.77) patients with events per 100 patient-years in the 11 mg prolonged-release tablet once daily and 5 mg film-coated tablet twice daily groups, respectively. The unadjusted hazard ratio was 1.30 (95% CI: 0.67, 2.50) at 12 months and 1.93 (95% CI: 1.15, 3.24) at 36 months for the 11 mg prolonged-release once daily dose compared to the 5 mg film-coated twice daily dose. Data is based on a small number of patients with events observed with relatively large confidence intervals and limited follow up time.
Viral reactivation
Patients treated with tofacitinib who are Japanese or Korean, or patients with long standing RA who have previously received two or more biological DMARDs, or patients with an ALC less than 1,000 cells/mm³, or patients treated with 10 mg twice daily may have an increased risk of herpes zoster (see section 4.4).
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years or older with at least one additional cardiovascular risk factor, an increase in herpes zoster events was observed in patients treated with tofacitinib compared to TNF inhibitors. The incidence rates (95% CI) for herpes zoster for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors were 3.75 (3.22, 4.34), 3.94 (3.38, 4.57), and 1.18 (0.90, 1.52) patients with events per 100 patient-years, respectively.
Laboratory tests
Lymphocytes
In the controlled RA clinical studies, confirmed decreases in ALC below 500 cells/mm³ occurred in 0.3% of patients and for ALC between 500 and 750 cells/mm³ in 1.9% of patients for the 5 mg twice daily and 10 mg twice daily doses combined.
In the RA long-term safety population, confirmed decreases in ALC below 500 cells/mm 3 occurred in 1.3% of patients and for ALC between 500 and 750 cells/mm³ in 8.4% of patients for the 5 mg twice daily and 10 mg twice daily doses combined.
Confirmed ALC less than 750 cells/mm³ were associated with an increased incidence of serious infections (see section 4.4).
In the clinical studies in UC, changes in ALC observed with tofacitinib treatment were similar to the changes observed in clinical studies in RA.
Neutrophils
In the controlled RA clinical studies, confirmed decreases in ANC below 1,000 cells/mm³ occurred in 0.08% of patients for the 5 mg twice daily and 10 mg twice daily doses combined. There were no confirmed decreases in ANC below 500 cells/mm³ observed in any treatment group. There was no clear relationship between neutropenia and the occurrence of serious infections.
In the RA long-term safety population, the pattern and incidence of confirmed decreases in ANC remained consistent with what was seen in the controlled clinical studies (see section 4.4).
In the clinical studies in UC, changes in ANC observed with tofacitinib treatment were similar to the changes observed in clinical studies in RA.
η4. Platelets
Patients in the Phase 3 controlled clinical studies (RA, PsA, AS, UC) were required to have a platelet count ≥100,000 cells/mm³ to be eligible for enrolment, therefore, there is no information available for patients with a platelet count <100,000 cells/mm³ before starting treatment with tofacitinib.
Liver enzyme tests
Confirmed increases in liver enzymes greater than 3 times the upper limit of normal (3x ULN) were uncommonly observed in RA patients. In those patients experiencing liver enzyme elevation, modification of treatment regimen, such as reduction in the dose of concomitant DMARD, interruption of tofacitinib, or reduction in tofacitinib dose, resulted in decrease or normalisation of liver enzymes.
In the controlled portion of the RA phase 3 monotherapy study (0-3 months) (study I, see section 5.1), ALT elevations greater than 3x ULN were observed in 1.65%, 0.41%, and 0% of patients receiving placebo, tofacitinib 5 mg and 10 mg twice daily, respectively. In this study, AST elevations greater than 3x ULN were observed in 1.65%, 0.41% and 0% of patients receiving placebo, tofacitinib 5 mg and 10 mg twice daily, respectively.
In the RA phase 3 monotherapy study (0-24 months) (study VI, see section 5.1), ALT elevations greater than 3x ULN were observed in 7.1%, 3.0%, and 3.0% of patients receiving MTX, tofacitinib 5 mg and 10 mg twice daily, respectively. In this study, AST elevations greater than 3x ULN were observed in 3.3%, 1.6% and 1.5% of patients receiving MTX, tofacitinib 5 mg and 10 mg twice daily, respectively.
In the controlled portion of the RA phase 3 studies on background DMARDs (0-3 months) (studies II-V, see section 5.1), ALT elevations greater than 3x ULN were observed in 0.9%, 1.24% and 1.14% of patients receiving placebo, tofacitinib 5 mg and 10 mg twice daily, respectively. In these studies, AST elevations greater than 3x ULN were observed in 0.72%, 0.5% and 0.31% of patients receiving placebo, tofacitinib 5 mg and 10 mg twice daily, respectively.
In the RA long-term extension studies, on monotherapy, ALT elevations greater than 3x ULN were observed in 1.1% and 1.4% of patients receiving tofacitinib 5 mg and 10 mg twice daily, respectively. AST elevations greater than 3x ULN were observed in <1.0% in both the tofacitinib 5 mg and 10 mg twice daily groups.
In the RA long-term extension studies, on background DMARDs, ALT elevations greater than 3x ULN were observed in 1.8% and 1.6% of patients receiving tofacitinib 5 mg and 10 mg twice daily, respectively. AST elevations greater than 3x ULN were observed in <1.0% in both the tofacitinib 5 mg and 10 mg twice daily groups.
In the RA long-term extension studies, on background DMARDs, ALT elevations greater than 3x ULN were observed in 1.8% and 1.6% of patients receiving tofacitinib 5 mg and 10 mg twice daily, respectively. AST elevations greater than 3x ULN were observed in <1.0% in both the tofacitinib 5 mg and 10 mg twice daily groups.
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years or older with at least one additional cardiovascular risk factor, ALT elevations greater than or equal to 3x ULN were observed in 6.01%, 6.54% and 3.77% of patients receiving tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors respectively. AST elevations greater than or equal to 3 x ULN were observed in 3.21%, 4.57% and 2.38% of patients receiving tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors respectively.
In the clinical studies in UC, changes in liver enzyme tests observed with tofacitinib treatment were similar to the changes observed in clinical studies in RA.
Lipids
Elevations in lipid parameters (total cholesterol, LDL cholesterol, HDL cholesterol, triglycerides) were first assessed at 1 month following initiation of tofacitinib in the controlled double-blind clinical trials of RA. Increases were observed at this time point and remained stable thereafter.
Changes in lipid parameters from baseline through the end of the study (6-24 months) in the controlled clinical studies in RA are summarised below:
- Mean LDL cholesterol increased by 15% in the tofacitinib 5 mg twice daily arm and 20% in the tofacitinib 10 mg twice daily arm at month 12, and increased by 16% in the tofacitinib 5 mg twice daily arm and 19% in the tofacitinib 10 mg twice daily arm at month 24.
- Mean HDL cholesterol increased by 17% in the tofacitinib 5 mg twice daily arm and 18% in the tofacitinib 10 mg twice daily arm at month 12, and increased by 19% in the tofacitinib 5 mg twice daily arm and 20% in the tofacitinib 10 mg twice daily arm at month 24.
Upon withdrawal of tofacitinib treatment, lipid levels returned to baseline.
Mean LDL cholesterol/HDL cholesterol ratios and Apolipoprotein B (ApoB)/ApoA1 ratios were essentially unchanged in tofacitinib-treated patients.
In an RA controlled clinical trial, elevations in LDL cholesterol and ApoB decreased to pretreatment levels in response to statin therapy.
In the RA long-term safety populations, elevations in the lipid parameters remained consistent with what was seen in the controlled clinical studies.
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years or older with at least one additional cardiovascular risk factor, changes in lipid parameters from baseline through 24 months are summarised below:
- Mean LDL cholesterol increased by 13.80%, 17.04%, and 5.50% in patients receiving tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitor, respectively, at month 12. At month 24, the increase was 12.71%, 18.14%, and 3.64%, respectively,
- Mean HDL cholesterol increased by 11.71%, 13.63%, and 2.82% in patients receiving tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitor, respectively, at month 12. At month 24, the increase was 11.58%, 13.54%, and 1.42%, respectively.
In the clinical studies in UC, changes in lipids observed with tofacitinib treatment were similar to the
changes observed in clinical studies in RA.
Myocardial infarction
Rheumatoid arthritis
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, the incidence rates (95% CI) for non-fatal myocardial infarction for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors were 0.37 (0.22, 0.57), 0.33 (0.19, 0.53), and 0.16 (0.07, 0.31) patients with events per 100 patient-years, respectively. Few fatal myocardial infarctions were reported with rates similar in patients treated with tofacitinib compared to TNF inhibitors (see sections 4.4 and 5.1). The study required at least 1500 patients to be followed for 3 years.
Malignancies excluding NMSC
Rheumatoid arthritis
In a large (N=4,362) randomised post-authorisation safety study in patients with RA who were 50 years of age or older with at least one additional cardiovascular risk factor, the incidence rates (95% CI) for lung cancer for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors were 0.23 (0.12, 0.40), 0.32 (0.18, 0.51), and 0.13 (0.05, 0.26) patients with events per 100 patient-years, respectively (see sections 4.4 and 5.1). The study required at least 1500 patients to be followed for 3 years.
The incidence rates (95% CI) for lymphoma for tofacitinib 5 mg twice daily, tofacitinib 10 mg twice daily, and TNF inhibitors were 0.07 (0.02, 0.18), 0.11 (0.04, 0.24), and 0.02 (0.00, 0.10) patients with events per 100 patient-years, respectively (see sections 4.4 and 5.1).
Paediatric population
Polyarticular juvenile idiopathic arthritis and juvenile PsA
The adverse reactions in JIA patients in the clinical development program were consistent in type and frequency with those seen in adult RA patients, with the exception of some infections (influenza, pharyngitis, sinusitis, viral infection) and gastrointestinal or general disorders (abdominal pain, nausea, vomiting, pyrexia, headache, cough), which were more common in JIA paediatric population. MTX was the most frequent concomitant csDMARD used (on Day 1, 156 of 157 patients on csDMARDs took MTX). There are insufficient data regarding the safety profile of tofacitinib used concomitantly with any other csDMARDs.
Infections
In the double-blind portion of the pivotal Phase 3 trial (Study JIA-I), infection was the most commonly reported adverse reaction (44.3%). The infections were generally mild to moderate in severity.
In the integrated safety population, 7 patients had serious infections during treatment with tofacitinib within the reporting period (up to 28 days after the last dose of study medication), representing an incidence rate of 1.92 patients with events per 100 patient-years: pneumonia, epidural empyema (with sinusitis and subperiosteal abscess), pilonidal cyst, appendicitis, escherichia pyelonephritis, abscess limb, and UTI.
In the integrated safety population, 3 patients had non-serious events of herpes zoster within the reporting window representing an incidence rate of 0.82 patients with events per 100 patient-years. One (1) additional patient had an event of serious HZ outside the reporting window.
Hepatic events
Patients in the JIA pivotal study were required to have AST and ALT levels less than 1.5 times the upper limit of normal to be eligible for enrolment. In the integrated safety population, there were 2 patients with ALT elevations ≥3 times the ULN at 2 consecutive visits. Neither event met Hy's Law criteria. Both patients were on background MTX therapy and each event resolved after discontinuation of MTX and permanent discontinuation of tofacitinib.
Laboratory tests
Changes in laboratory tests in JIA patients in the clinical development program were consistent with those seen in adult RA patients. Patients in the JIA pivotal study were required to have a platelet count ≥100,000 cells/mm³ to be eligible for enrolment, therefore, there is no information available for JIA patients with a platelet count <100,000 cells/mm³ before starting treatment with tofacitinib.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.