YORVIPATH Solution for injection Ref.[51687] Active ingredients: Palopegteriparatide
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Ascendis Pharma Bone Diseases A/S, Tuborg Boulevard 12, DK-2900 Hellerup, Denmark
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Calcium homeostasis, parathyroid hormones and analogues
ATC code: H05AA05
Mechanism of action
Endogenous parathyroid hormone (PTH) is secreted by the parathyroid glands as a polypeptide of 84 amino acids. PTH exerts its action via cell-surface parathyroid hormone receptors, for example, expressed in bone, kidney and nerve tissue. Activation of PTH1R stimulates bone turnover, increases renal calcium reabsorption and phosphate excretion and facilitates synthesis of active vitamin D.
Palopegteriparatide is a prodrug, consisting of PTH(1-34) conjugated to a methoxypolyethylene glycol carrier (mPEG) via a proprietary TransCon Linker. PTH(1-34) and its main metabolite, PTH(1-33), have similar affinity to and activation of PTH1R as endogenous PTH. At physiological conditions, PTH is cleaved from palopegteriparatide in a controlled manner to provide a continuous systemic exposure of active PTH.
Clinical efficacy and safety
Study in patients with established hypoparathyroidism
The pivotal phase 3 PaTHway clinical trial (TCP-304) assessed the efficacy and safety of Yorvipath as PTH replacement therapy for adults with hypoparathyroidism. The 26‑week double-blind, placebo-controlled period of the clinical trial included patients randomised (3:1) to Yorvipath at a starting dose of 18 micrograms/day or placebo, co-administered with conventional therapy (calcium supplement and active vitamin D). Randomisation was stratified by aetiology of hypoparathyroidism (i.e., postsurgical vs. all other causes). Study treatment (palopegteriparatide or placebo) and conventional therapy were subsequently titrated according to a dosing algorithm guided by albumin-adjusted serum calcium levels.
Patients' mean age at recruitment was 49 years (19 to 78 years of age; 12% were ≥65 years old), and the majority of patients were female (78%) and Caucasian (93%). Eighty-five percent (85%) of patients had hypoparathyroidism acquired from neck surgery. Of the patients with other aetiologies of hypoparathyroidism, 7 (8.5%) patients had idiopathic disease, 2 had autoimmune polyglandular syndrome type 1 (APS-1), 1 had autosomal dominant hypocalcaemia type 1 (ADH1, CaSR mutation), 1 had DiGeorge Syndrome, and 1 had hypoparathyroidism, sensorineural deafness and renal dysplasia (HDR) syndrome (GATA3 mutation).
Prior to randomisation, all patients underwent an approximate 4-week screening period in which calcium and active vitamin D supplements were adjusted to achieve an albumin-adjusted serum calcium concentration between 1.95 to 2.64 mmol/L (7.8 to 10.6 mg/dL), a magnesium concentration ≥0.53 mmol/L (≥1.3 mg/dL) and below the upper reference range of normal, and a 25(OH) vitamin D concentration between 50 to 200 nmol/L (20 to 80 ng/mL). For conventional therapy, patients were treated with mean baseline doses of calcium (elemental) of 1 839 mg/day. Mean baseline doses of active vitamin D were 0.75 micrograms/day in calcitriol-treated patients (n=70), and 2.3 micrograms/day in alfacalcidol-treated patients (n=12). Baseline mean albumin-adjusted serum calcium and mean 24-hour urine calcium were similar in both treatment groups: mean serum calcium was 2.2 mmol/L (8.8 mg/dL) and 2.15 mmol/L (8.6 mg/dL) and mean 24-hour urine calcium was 392 mg/day and 329 mg/day, for Yorvipath and placebo, respectively.
Primary endpoint
The composite primary efficacy endpoint was defined as the proportion of patients at week 26 who achieved: serum calcium levels in the normal range (2.07 to 2.64 mmol/L [8.3 to 10.6 mg/dL]), independence from conventional therapy defined as requiring no active vitamin D and ≤600 mg/day of calcium supplementation, and no increase in prescribed study treatment within 4 weeks prior to week 26. Key secondary endpoints included a subset of Hypoparathyroidism Patient Experience Scale (HPES) domain scores and 36-Item Short Form Survey (SF-36) subscale scores.
The number of patients meeting the composite primary endpoint compared with the placebo group and each component of the primary endpoint at week 26 is presented in table 3.
Table 3. TCP-304: Response rate based on primary endpoint at week 26:
| Yorvipath (N=61) (n, %) | Placebo (N=21) (n, %) | Response rate difference (95% CI) | |
|---|---|---|---|
| Response at week 26 | 48 (78.7%) | 1 (4.8%) | 74.0% (60.4%, 87.6%) p<0.0001 |
| Response for each component | |||
| Albumin-adjusted serum calcium within normal rangea | 49 (80.3%) | 10 (47.6%) | 32.7% (9.2%, 56.3%) |
| Independence from active vitamin Db | 60 (98.4%) | 5 (23.8%) | 74.6% (56.1%, 93.1%) |
| Independence from therapeutic doses of calciumc | 57 (93.4%) | 1 (4.8%) | 88.7% (77.7%, 99.7%) |
| No dose increase in Yorvipathd | 57 (93.4%) | 12 (57.1%) | 36.4% (14.2%, 58.5%) |
a The normal range for albumin-adjusted serum calcium was 2.07 to 2.64 mmol/L (8.3 to 10.6 mg/dL).
b All daily standing doses of active vitamin D equal to zero AND use of PRN doses for ≤7 days within 4 weeks prior to week 26 visit.
c Average daily standing doses of elemental calcium ≤600 mg AND use of PRN doses on ≤7 days within 4 weeks prior to week 26 visit.
d No dose increase in Yorvipath within 4 weeks prior to week 26 visit.
Abbreviations: CI: confidence interval; PRN: pro re nata.
Secondary endpoints
Conventional therapy intake: calcium and active vitamin D doses
In the phase 3 PaTHway trial, at week 26, 93% (57/61) of patients in the Yorvipath group were able to discontinue conventional therapy (i.e., discontinue active vitamin D and therapeutic doses of calcium). All patients in the Yorvipath group discontinued active vitamin D by week 8 and had sustained reduction in therapeutic doses of calcium. There was a significant reduction in conventional therapy intake in the Yorvipath group from baseline to week 26 compared with placebo: active vitamin D (nominal p-value<0.0001), calcium dose (nominal p-value=0.0003), and daily pill burden (nominal p-value<0.0001) (table 4).
Table 4. Secondary endpoints: conventional therapy intake at week 26 - blinded period (ITT population):
| Yorvipath (n/N=60/61)a | Placebo (n/N=19/21)a | Nominal p-value | |||
|---|---|---|---|---|---|
| Baseline | Week 26 | Baseline | Week 26 | ||
| Supplemental active vitamin D dose (mcg), mean (SD) | 1.0 (0.7) | 0.0 (0.0) | 1.0 (0.6) | 0.6 (0.7) | <0.0001 |
| Supplemental calcium dose (mg), mean (SD) | 1 737 (907) | 274 (177) | 2 089 (1 448) | 1 847 (1 326) | 0.0003 |
| Daily pill burden (number of conventional therapy pills), mean (SD) | 6.6 (2.1) | 0.5 (1.7) | 6.3 (2.8) | 5.4 (3.2) | <0.0001 |
Nominal p-value from testing the differences in change from baseline to week 26 between Yorvipath and placebo.
a N is the number of patients in the ITT population; n is the number of patients with data at both baseline and week 26.
Serum biochemistries
Mean serum calcium initially increased and stayed within the normal range in palopegteriparatidetreated patients (figure 2). In placebo patients, serum calcium levels decreased slightly, falling below normal range at week 2 (mean observed value: 2.06 mmol/L) and at week 26 (mean observed value: 2.06 mmol/L). The LS mean treatment difference between Yorvipath and placebo was 0.17 mmol/L (95% CI: 0.100, 0.247; nominal p<0.0001) at week 26.
Figure 2. Serum calcium (mean ± SE) by visit - blinded period (ITT population):
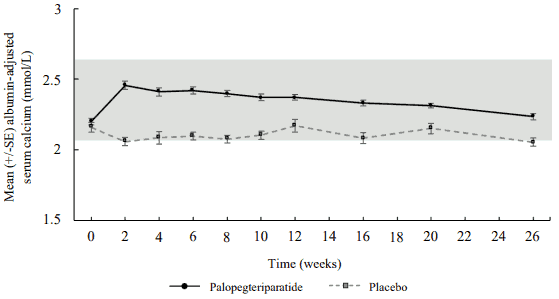
Mean serum phosphate levels for palopegteriparatide-treated patients were in the normal range at baseline and fell within the normal range through week 26 (Mean change from baseline to week 26 was -0.13 mmol/L). Mean serum calcium x phosphate product decreased in patients treated with Yorvipath and remained stable within the normal range through week 26.
24-hour urine calcium excretion
Yorvipath therapy normalised mean 24-hour urine calcium excretion and showed greater reduction in 24-hour urine calcium versus placebo.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Yorvipath in one or more subsets of the paediatric population in hypoparathyroidism, as per paediatric investigation plan (PIP) decision, for the indication of treatment of hypoparathyroidism.
5.2. Pharmacokinetic properties
Absorption
Following daily subcutaneous administration, palopegteriparatide releases PTH via autocleavage of the TransCon Linker with first-order kinetics, resulting in continuous exposure over 24 hours within the estimated normal range (figure 3).
Figure 3. Mean released PTH* following subcutaneous administration of palopegteriparatide at steady state in patients with hypoparathyroidism:
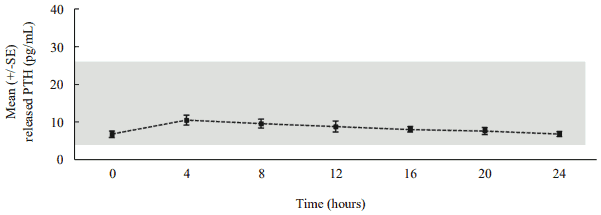
The estimated normal range for PTH(1-34) is approximately 4 to 26 pg/mL. This is calculated based on PTH(1-34) constituting 40% of the molecular weight of PTH(1-84)** and the normal range (10 to 65 pg/mL) for PTH(1-84).
* Mean palopegteriparatide dose (range): 22.3 (12-33) mcg PTH(1-34)/day, n=7, released PTH: sum of PTH(1-34) and PTH(1-33).
** PTH(1-84) = endogenous parathyroid hormone.
In patients with hypoparathyroidism administered palopegteriparatide corresponding to 18 mcg of PTH(1-34)/day, the predicted maximum plasma concentration (Cmax) (CV%) of palopegteriparatide was 5.18 ng/mL (36%) and the predicted Cmax (CV%) for released PTH was 6.9 pg/mL (22%) with a median time to reach maximum concentrations (Tmax) of 4 hours. The predicted exposure over the 24-hour dosing interval (area under the curve, AUC) (CV%) for released PTH was 150 pg*h/mL (22%).
Following multiple subcutaneous doses of palopegteriparatide in the range of 12 to 24 mcg PTH(1-34)/day, the palopegteriparatide and released PTH concentrations increased in a dose-proportional manner reaching steady-state within approximately 10 and 7 days, respectively. The peak-to-trough ratio was low, approximately 1.1 and 1.5 over 24 hours at steady state for palopegteriparatide and released PTH, respectively. Palopegteriparatide accumulated after multiple dosing by up to 18-fold for AUC.
Distribution
The apparent volume of distribution (CV%) of palopegteriparatide is estimated to 4.8 L (50%) and to 8.7 L (18%) for released PTH.
Biotransformation
PTH released from palopegteriparatide is composed of PTH(1-34) and the metabolite PTH(1-33). PTH is renally metabolised and cleared.
Elimination
In healthy adults, the clearance (CV%) of palopegteriparatide at steady state is estimated to be 0.58 L/day (52%) with a predicted half-life of 70 hours. The apparent half-life of PTH released from palopegteriparatide is approximately 60 hours. In the liver, most of the PTH is cleaved by cathepsins. In the kidney, a small amount of PTH binds to PTH1R, but most is excreted by glomerular filtration.
Pharmacokinetic/pharmacodynamic relationship
In a pharmacodynamic/pharmacokinetic sub-study in hypoparathyroid patients, daily subcutaneous administration of palopegteriparatide (mean dose (range): 22.3 (12-33) mcg PTH(1-34)/day) increased serum calcium levels to within the normal range (see figure 4). The increase in serum calcium levels occurred in a dose-related manner, supporting the ability to titrate palopegteriparatide according to measured serum calcium values in the individual patient.
Figure 4. Mean albumin-adjusted serum calcium concentrations following subcutaneous administration of palopegteriparatide at steady state in patients with hypoparathyroidism:
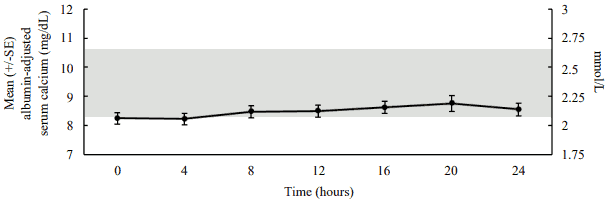
The normal range for albumin-adjusted serum calcium is 2.07 to 2.64 mmol/L (8.3 to 10.6 mg/dL) as denoted by the grey shading. Mean palopegteriparatide dose (range): 22.3 (12-33) mcg PTH(1-34)/day, n=7.
Special populations
The pharmacokinetics of released PTH was not influenced by sex or body weight. The data for race and ethnicity did not show any trends indicating differences, but the available data are too limited to make definitive conclusions.
Elderly
The pharmacokinetics of released PTH was not influenced by age (19 to 76 years old).
Renal impairment
Yorvipath has been administered to patients with hypoparathyroidism with an eGFR of ≥30 mL/min in long-term clinical trials without the need for dose adjustment beyond the trial titration algorithm. No clinical trials were conducted in patients with hypoparathyroidism with severe renal impairment (<30 mL/min) or on dialysis. In a trial where Yorvipath was administered as a single dose to non-hypoparathyroid subjects with renal impairment, palopegteriparatide exposure and resulting serum calcium levels were similar in subjects with mild, moderate, and severe renal impairment as compared to subjects without renal impairment.
5.3. Preclinical safety data
No special hazard for humans were revealed in the conventional studies of safety pharmacology, genotoxicity, and local tolerance conducted with palopegteriparatide.
At the highest dose levels in all animal species employed, repeated dosing resulted in adverse persistent hypercalcemia, which in some studies led to premature death/euthanasia, clinical signs, body weight loss and/or soft tissue mineralisation observed mainly in the kidneys. These findings are considered results of persistent exaggerated PTH pharmacology and of no relevance in a clinical setting where dose adjustments are performed to ensure normalised serum calcium.
In accordance with the expected pharmacological effects, repeated daily administration of palopegteriparatide increased bone turnover in rats. At low dose levels (2-fold the maximum recommended human dose (MRHD), based on exposure to released PTH by AUC) in rats, the increased bone turnover induced overall net catabolic bone effects. At high dose levels (5-fold the MRHD, based on exposure to released PTH by AUC) in rats, the increased bone turnover resulted in a net anabolic bone effect. Physeal dysplasia was observed at the highest dose level (9-fold the MRHD, based on exposure to released PTH by AUC) in rats. These effects are of no relevance in a clinical setting where Yorvipath doses are individually adjusted.
There were no cardiovascular findings in monkeys up to and including the highest dose tested in single- (3-fold the MRHD, based on exposure to released PTH by Cmax) or repeat-dose studies (0.98-fold the MRHD, based on exposure to released PTH by Cmax).
Increased occurrence of osteosarcomas has been observed in carcinogenicity studies with short-lived PTH analogues in rats, but there is no evidence of increased risk of osteosarcoma in patients treated with short-lived PTH analogues. No carcinogenicity study has been conducted with palopegteriparatide.
In animal reproduction studies, administration of palopegteriparatide to pregnant rats and rabbits during the period of organogenesis resulted in no evidence of embryo-lethality, foetotoxicity or dysmorphogenesis up to and including the highest doses tested (8- and 7-fold, respectively, the MRHD, based on exposure to released PTH by AUC). Exaggerated PTH pharmacological effects were observed at the highest doses tested in the pregnant rats and rabbits (increased serum calcium levels, decreased body weight, decreased food consumption and/or clinical signs). The exposures at the no observed adverse effect level (NOAEL) for maternal toxicity were 2- and 3-fold the MRHD, based on exposure to released PTH by AUC in pregnant rats and rabbits, respectively. A pre- and postnatal developmental study has not been conducted with palopegteriparatide.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.