ZEPZELCA Powder for concentrate for solution for infusion Ref.[116643] Active ingredients: Lurbinectedin
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Pharma Mar, S.A., Avda. de los Reyes 1, Polígono Industrial La Mina, 28770 Colmenar Viejo (Madrid), Spain, Tel: +34 91 846 60 00, Fax: +34 91 846 60 01
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, other antineoplastic agents
ATC code: L01XX69
Mechanism of action
Lurbinectedin inhibits the oncogenic transcription process through (i) its binding to CG-rich sequences of DNA, located within promoters of protein-coding genes; (ii) the eviction of oncogenic transcription factors from their binding sites; and (iii) the stalling of elongating RNA polymerase II and its specific degradation by the ubiquitin/proteasome machinery with all these processes leading to subsequent cell cycle arrest and tumour cell apoptosis.
Lurbinectedin suppresses the expression of inflammatory and motility-related genes at non-toxic nanomolar concentrations in vitro, while also inhibiting cell migration and adhesion. At higher concentrations, it induces apoptosis in monocytes and macrophages through caspase-8 activation. In vivo (murine models), antitumour dosing (0.18–0.20 mg/kg) restricts tumour growth, reduces specific immune cell populations, and decreases tumour vascularity.
Pharmacodynamic effects
Cardiac Electrophysiology
The potential for QTc prolongation with lurbinectedin was evaluated in 39 patients with advanced cancer. Large effects (>10 ms) on the QTc interval were not detected with lurbinectedin dosed at 3.2 mg/m² every 21 days.
Clinical efficacy and safety
Extensive-stage small cell lung cancer
The efficacy of maintenance treatment with ZEPZELCA in combination with atezolizumab was investigated in 483 patients with first-line ES-SCLC in IMforte, a randomised, multicentre, open-label study. Participants were eligible for randomisation if they have achieved CR, PR, or SD by RECIST v1.1 based on radiographic assessment within 28 days prior to randomisation after completion of 4 cycles of induction treatment with atezolizumab, carboplatin and etoposide and if they had an ECOG performance status of 0 or 1. Eligible patients were randomised 1:1 to receive maintenance treatment with either lurbinectedin with atezolizumab or atezolizumab alone. Unless contraindicated, primary prophylaxis with G-CSF was given for patients assigned to the lurbinectedin with atezolizumab arm. The study excluded patients with CNS metastases, a history of autoimmune disease, or administration of systemic immunosuppressive medicines within 1 week prior to enrolment. Randomisation was stratified by ECOG performance status (0 vs. 1), lactate dehydrogenase (LDH) (≤ ULN vs. > ULN), presence of liver metastases at enrolment (yes vs. no), and prior receipt of prophylactic cranial irradiation (yes vs. no).
Patients were randomised to one of the following two treatment arms:
- ZEPZELCA 3.2 mg/m² intravenous with atezolizumab 1200 mg intravenous once every 3 weeks until disease progression or unacceptable toxicity, or
- Atezolizumab 1200 mg intravenous once every 3 weeks until disease progression or unacceptable toxicity.
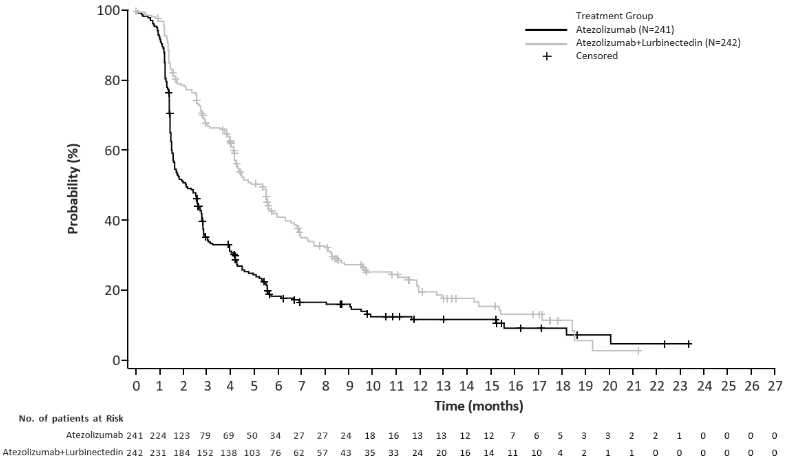
Primary efficacy endpoints were overall survival (OS) and Independent Review Facility (IRF)-assessed progression-free survival (PFS) per the Response Evaluation Criteria in Solid Tumours (RECIST) v1.1 in the randomised population. (see Table 5).
A total of 483 patients were randomised: 242 to the ZEPZELCA with atezolizumab arm and 241 to the atezolizumab arm. The median age was 66 years (range 35 to 85 years, being 13% ≥75 years). The majority of patients were White (81.6%); 12.8% were Asian, 6.6% were Hispanic and <1% were Black or African American. Most patients were male (62.5%) and 97.5% were current or previous smokers. Baseline ECOG performance status was 0 (42.9%) or 1 (57.1%).
Efficacy results are presented in Table 5 and Figures 1 and 2.
Table 4. Efficacy results from IMforte:
| lurbinectedin with atezolizumab N=242 | atezolizumab N=241 | |
| Overall Survival1 | ||
| Deaths (%) | 113 (46.7%) | 136 (56.4%) |
| Median, months | 13.2 | 10.6 |
| (95% CI) | (11.9, 16.4) | (9.5, 12.2) |
| Hazard ratio2 (95% CI) | 0.73 (0.57, 0.95) | |
| p-value3,6 | 0.0174 | |
| Progression-Free Survival1,4,5 | ||
| Number of events (%) | 174 (71.9%) | 202 (83.8%) |
| Median, months | 5.4 | 2.1 |
| (95% CI) | (4.2, 5.8) | (1.6, 2.7) |
| Hazard ratio2 (95% CI) | 0.54 (0.43, 0.67) | |
| p-value3,7 | <0.0001 | |
Cut-off: 29 July 2024
1 Measured from the time of randomisation
2 Stratified by ECOG performance status, LDH level, presence of liver metastases and prior receipt of prophylactic cranial irradiation
3 Based on the stratified log-rank test
4 As determined by IRF
5 per RECIST v1.1
6 Compared to the allocated alpha of 0.0313 (two-sided) for this interim OS analysis.
7 Compared to the allocated alpha of 0.001 (two-sided) for this final PFS analysis.
CI=confidence interval
Figure 1. Kaplan-Meier plot of overall survival in IMforte:
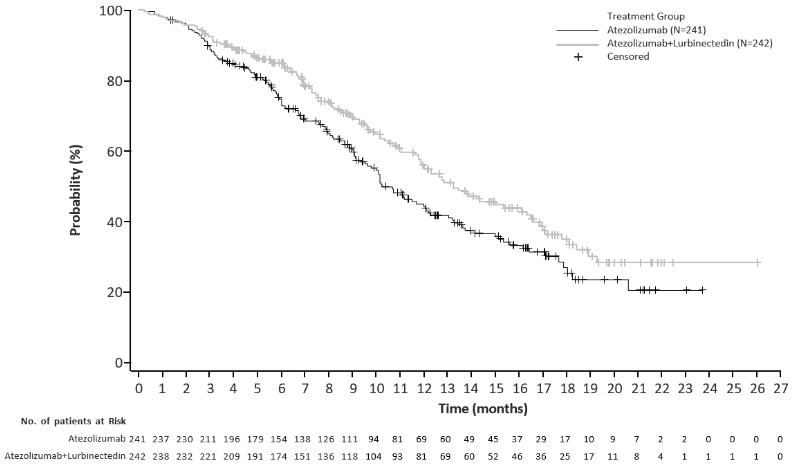
Figure 2. Kaplan-Meier plot of IRF-assessed progression free survival in IMforte:
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with ZEPZELCA in all subsets of the paediatric population in the treatment of SCLC (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
After a 3.2 mg/m² lurbinectedin dose administered as a 1-hour intravenous infusion, geometric means of total plasma Cmax and AUC∞, were 107 μg/L and 551 μg*h/L, respectively. No accumulation of lurbinectedin in plasma is observed upon repeated administrations every 21 days.
Distribution
Typical volume of distribution of lurbinectedin at steady state is 504 L. Binding to plasma proteins is approximately 99%, to both albumin and α-1-acid glycoprotein, with a calculated blood-to-plasma partitioning ratio of 0.68.
Biotransformation
In vitro studies
In vitro studies with human liver microsomes and supersomes indicate that CYP3A4 is the main CYP enzyme responsible for the hepatic metabolism of lurbinectedin.
Cytochrome P450 (CYP) Enzymes: Lurbinectedin is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, or CYP3A4. Lurbinectedin is not an inducer of CYP1A2 or CYP3A4. The potential of lurbinectedin to induce CYP2B6 is unknown.
Transporter Systems: Lurbinectedin is a substrate of MDR1 (P-gp), but is not a substrate of OATB1P1, OATP1B3, OCT1, or MATE1. In vitro, lurbinectedin showed inhibitory potential towards MDR1, OATP1B1, OATP1B3, and OCT1 however, these findings are not considered clinically relevant. Lurbinectedin is not an inhibitor of BCRP, BSEP, MATE1, OAT1, OAT3, or OCT2.
Elimination
The terminal half-life of lurbinectedin is 51 hours. Total plasma clearance of lurbinectedin is 11 L/h.
The major route of lurbinectedin-related radioactivity excretion was via faeces (89% of dose), with only traces amounts of unchanged lurbinectedin detected in faeces (<0.2% of dose). Excretion in urine was the minor route (6% of dose), mainly as unchanged compound (1% of dose) and one metabolite (up to 1% of dose).
Linearity/non-linearity
Lurbinectedin pharmacokinetics is linear at the dose range of 0.02-6.9 mg/m².
Special populations
Population pharmacokinetics analyses showed that weight (range: 39-154 kg), age (range: 18-85 years), and gender do not have a clinically meaningful influence on the systemic exposure of lurbinectedin.
Hepatic impairment
A dedicated study was conducted to evaluate the influence of varying degrees of hepatic impairment (HI) on lurbinectedin in patients with advanced solid tumours. Patients were classified according to the National Cancer Institute Organ Dysfunction Working Group (NCI-ODWG) classification as having normal hepatic function or mild (total bilirubin ≤ ULN and AST > ULN, or total bilirubin >1 to ≤1.5 × ULN and AST = any), moderate (total bilirubin >1.5 to ≤3 × ULN and AST = any), or severe (total bilirubin >3 × ULN) HI. Patients with normal hepatic function and mild HI received lurbinectedin at 3.2 mg/m² and patients with moderate and severe HI received lurbinectedin at 1.6 mg/m². No statistically significant differences were observed on total lurbinectedin pharmacokinetics among cohorts. A statistically significant higher dose-normalised M1 AUC metabolite/parent ratio (MPR) was observed in patients with severe HI (ratio: 5.95, 90% CI: 2.54–13.98) and moderate HI (ratio: 8.65, 90% CI: 3.94–19.01) compared to patients with mild HI. No statistically significant differences were observed in M4 MPR according to HI groups.
Based on population pharmacokinetic analysis, no apparent pharmacokinetic difference was observed in 125 patients with mild who received lurbinectedin 3.2 mg/m² every 21 days as compared to 625 patients with normal hepatic function.
Renal impairment
No dedicated studies of lurbinectedin have been conducted in patients with renal impairment. Based on population pharmacokinetic analyses, no apparent pharmacokinetic difference was observed in 165 patients with mild renal impairment (CrCL of 60-89 mL/min), 73 patients with moderate renal impairment (CrCL of 30-59 mL/min), and one patient with severe renal impairment (CrCL of 26 mL/min) who received lurbinectedin 3.2 mg/m² every 21 days as compared to 166 patients with normal renal function. The pharmacokinetic characteristics of lurbinectedin in patients with CrCL <30 mL/min or patients on dialysis are unknown.
5.3. Preclinical safety data
Toxicology
The primary target for toxicity identified in nonclinical species (rat, dog and NHP) was characterised by severe, reversible and non-cumulative atrophy of bone marrow, which was associated with dose- related leukopenia, as well as thrombocytopenia and anaemia. In addition, lurbinectedin-treated animals experienced liver abnormalities (multiple dark areas or swollen liver, increased liver function markers, bile duct damage with necrosis and/or oedema, and hepatocellular degeneration/apoptosis and periportal hepatocytic hypertrophy). Additional findings were located in the gastro-intestinal tract (mucosal atrophy), kidneys (cortical tubular degeneration and vacuolation), heart (focal, slight to moderate myocardial degeneration and/or necrosis) and injection site (perivascular/vascular inflammatory reactions). A full recovery, after cessation of dosing, was noted for the majority of these alterations.
Genotoxicity
Positive genotoxicity results were obtained in vitro in mammalian cell lines showing dose related toxicity at all concentrations tested (range from 48 to 0.188 ng/mL). Positive genotoxicity findings are expected for lurbinectedin as a DNA-interacting antineoplastic agent (see section 4.6).
Carcinogenic potential
Carcinogenicity testing of lurbinectedin has not been performed.
Reproduction and development
Lurbinectedin induced maternal toxicity at the single dose MTD level of 0.6 mg/m² administered on Day 10 post-coitum and severe embryo-toxicity, leading to 100% embryo lethality (see section 4.4 and 4.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.