ZORVOLEX Capsule Ref.[50658] Active ingredients: Diclofenac
Source: FDA, National Drug Code (US) Revision Year: 2021
12.1. Mechanism of Action
Diclofenac has analgesic, anti-inflammatory, and antipyretic properties.
The mechanism of action of ZORVOLEX, like that of other NSAIDs, is not completely understood but involves inhibition of cyclooxygenase (COX-1 and COX-2).
Diclofenac is a potent inhibitor of prostaglandin synthesis in vitro. Diclofenac concentrations reached during therapy have produced in vivo effects. Prostaglandins sensitize afferent nerves and potentiate the action of bradykinin in inducing pain in animal models. Prostaglandins are mediators of inflammation. Because diclofenac is an inhibitor of prostaglandin synthesis, its mode of action may be due to a decrease of prostaglandins in peripheral tissues.
12.3. Pharmacokinetics
The relative bioavailability of ZORVOLEX 35 mg capsules was compared to diclofenac potassium immediate-release (IR) tablets 50 mg in 39 healthy subjects under fasted and fed conditions in a single-dose crossover study.
ZORVOLEX 35 mg capsules do not result in an equivalent systemic exposure to 50 mg diclofenac potassium IR tablets.
When taken under fasted conditions, a 20% lower dose of diclofenac in ZORVOLEX capsules resulted in a 23% lower mean systemic exposure (AUCinf) and a 26% lower mean peak concentration (Cmax) compared to diclofenac potassium IR tablets. The time to reach peak concentration (Tmax) was similar for ZORVOLEX and diclofenac potassium IR tablets and was ~1 hour for both.
When taken under fed conditions, a 20% lower dose of diclofenac in ZORVOLEX capsules resulted in a 23% lower mean systemic exposure (AUCinf) and a 48% lower mean Cmax compared to diclofenac potassium IR tablets. The Tmax for ZORVOLEX was delayed by approximately 1 hour compared to diclofenac potassium IR tablets (3.32 hours vs. 2.33 hours, respectively).
When taken under fed conditions, ZORVOLEX capsules resulted in an 11% lower mean systemic exposure (AUCinf) and a 60% lower mean Cmax compared to fasted conditions. Whereas diclofenac potassium IR tablets under fed conditions resulted in 8% - 10% lower mean systemic exposure (AUCinf) and 28% - 43% lower mean Cmax compared to fasted conditions, based on the results from two individual food effect studies. The Tmax for ZORVOLEX was delayed by approximately 2.32 hours under fed conditions compared to fasted conditions (3.32 hours vs. 1.00 hour, respectively), while the Tmax for diclofenac potassium IR tablets was delayed by approximately 1.00 - 1.33 hours under fed conditions compared to fasted conditions (1.70 vs. 0.74 hours and 2.33 vs. 1.00 hours, respectively in two studies).
There were no differences in elimination half-life between ZORVOLEX and diclofenac potassium IR tablets under fasted or fed conditions.
Absorption
Diclofenac is 100% absorbed after oral administration compared to IV administration as measured by urine recovery. However, due to first-pass metabolism, only about 50% of the absorbed dose is systemically available. After repeated oral administration, no accumulation of diclofenac in plasma occurred.
Administration of ZORVOLEX capsules 18 mg and 35 mg was associated with dose proportional pharmacokinetics.
Taking ZORVOLEX with food causes a significant decrease in the rate but not the overall extent of systemic absorption of diclofenac compared with taking ZORVOLEX on an empty stomach. ZORVOLEX capsules results in 60% lower Cmax, 11% lower AUCinf, and 2.32 hours delayed Tmax (1.0 hour during fasted versus 3.32 hours during fed) under the fed condition compared to the fasted condition. The effectiveness of ZORVOLEX when taken with food has not been studied in clinical studies. The decreased Cmax may be associated with decreased effectiveness. Taking ZORVOLEX with food may cause a reduction in effectiveness compared to taking ZORVOLEX on an empty stomach.
Distribution
The apparent volume of distribution (V/F) of diclofenac potassium is 1.3 L/kg. Diclofenac is more than 99% bound to human serum proteins, primarily to albumin. Serum protein binding is constant over the concentration range (0.15-105 mg/mL) achieved with recommended doses.
Diclofenac diffuses into and out of the synovial fluid. Diffusion into the joint occurs when plasma levels are higher than those in the synovial fluid, after which the process reverses and synovial fluid levels are higher than plasma levels. It is not known whether diffusion into the joint plays a role in the effectiveness of diclofenac.
Elimination
Diclofenac is eliminated through metabolism and subsequent urinary and biliary excretion of the glucuronide and the sulfate conjugates of the metabolites. The terminal half-life of unchanged diclofenac is approximately 2 hours.
Metabolism
Five diclofenac metabolites have been identified in human plasma and urine. The metabolites include 4'-hydroxy-, 5-hydroxy-, 3'-hydroxy-, 4',5-dihydroxy- and 3'-hydroxy-4'-methoxy diclofenac. The major diclofenac metabolite, 4'-hydroxy-diclofenac, has very weak pharmacologic activity. The formation of 4'-hydroxy-diclofenac is primarily mediated by CYP2C9. Both diclofenac and its oxidative metabolites undergo glucuronidation or sulfation followed by biliary excretion. Acylglucuronidation mediated by UGT2B7 and oxidation mediated by CYP2C8 may also play a role in diclofenac metabolism. CYP3A4 is responsible for the formation of minor metabolites, 5-hydroxy and 3'-hydroxy-diclofenac. In patients with renal dysfunction, peak concentrations of metabolites 4'-hydroxy and 5-hydroxy-diclofenac were approximately 50% and 4% of the parent compound after single oral dosing compared to 27% and 1% in normal healthy subjects.
Excretion
Diclofenac is eliminated through metabolism and subsequent urinary and biliary excretion of the glucuronide and the sulfate conjugates of the metabolites. Little or no free unchanged diclofenac is excreted in the urine. Approximately 65% of the dose is excreted in the urine, and approximately 35% in the bile as conjugates of unchanged diclofenac plus metabolites. Because renal elimination is not a significant pathway of elimination for unchanged diclofenac, dosing adjustment in patients with mild to moderate renal dysfunction is not necessary. The terminal half-life of unchanged diclofenac is approximately 2 hours.
Specific Populations
Pediatric
The pharmacokinetics of ZORVOLEX has not been investigated in pediatric patients.
Race
Pharmacokinetic differences due to race/ethnicity have not been identified.
Hepatic Impairment
No dedicated diclofenac pharmacokinetics studies in patients with hepatic impairment were conducted. Hepatic metabolism accounts for almost 100% of diclofenac elimination. Therefore, in patients with hepatic impairment, start with the lowest dose and if efficacy is not achieved, consider use of an alternate product [see Warnings and Precautions (5.3)].
Renal Impairment
Diclofenac pharmacokinetics has been investigated in subjects with renal insufficiency. No differences in the pharmacokinetics of diclofenac have been detected in studies of patients with renal impairment. In patients with renal impairment (inulin clearance 60-90, 30-60, and less than 30 mL/min; N=6 in each group), AUC values and elimination rate were comparable to those in healthy subjects [see Warnings and Precautions (5.6)].
Drug Interaction Studies
Aspirin
When NSAIDs were administered with aspirin, the protein binding of NSAIDs were reduced, although the clearance of free NSAID was not altered. The clinical significance of this interaction is not known. See Table 4 for clinically significant drug interactions of NSAIDs with aspirin [see Drug Interactions (7)].
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term carcinogenicity studies in rats given diclofenac sodium up to 2 mg/kg/day (approximately 0.2 times the maximum recommended human dose [MRHD] of ZORVOLEX based on body surface area [BSA] comparison) have revealed no significant increase in tumor incidence. A 2-year carcinogenicity study conducted in mice employing diclofenac sodium at doses up to 0.3 mg/kg/day (approximately 0.014 times the MRHD based on BSA comparison) in males and 1 mg/kg/day (approximately 0.04 times the MRHD based on BSA comparison) in females did not reveal any oncogenic potential.
Mutagenesis
Diclofenac sodium did not show mutagenic activity in in vitro point mutation assays in mammalian (mouse lymphoma) and microbial (yeast, Ames) test systems and was nonmutagenic in several mammalian in vitro and in vivo tests, including dominant lethal and male germinal epithelial chromosomal aberration studies in Chinese hamsters.
Impairment of Fertility
Diclofenac sodium administered to male and female rats at 4 mg/kg/day (approximately 0.4 times the MRHD based on BSA comparison) did not affect fertility.
14. Clinical Studies
Acute Pain
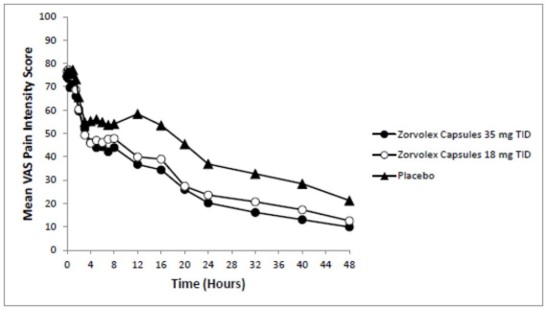
The efficacy of ZORVOLEX in the management of acute pain was demonstrated in a single multicenter, randomized, double-blind, placebo-controlled, parallel arm study comparing ZORVOLEX 18 mg and 35 mg taken three times a day, placebo, and celecoxib in patients with pain following bunionectomy. The study enrolled 428 patients with a mean age of 40 years (range 18 to 65 years) and a minimum pain intensity rating of at least 40 mm on a 100-mm visual analog scale (VAS) during the 9-hour period after discontinuation of the anesthetic block following bunionectomy surgery. Patients were randomized equally across the treatment groups.
The mean and range (in parenthesis) of pain intensities on the VAS at baseline were 74 mm (44 to 100 mm), 77 mm (41 to 100 mm), and 76 mm (40 to 100 mm) for the ZORVOLEX 35 mg, ZORVOLEX 18 mg, and placebo groups, respectively. One tablet of hydrocodone/acetaminophen 10 mg/325 mg was permitted every 4 to 6 hours as rescue medication. About 82% of patients in the ZORVOLEX 35 mg group, 85% of the patients in the ZORVOLEX 18 mg group, and 97% of patients in the placebo group took rescue medication for pain management during the study.
The average pain intensities over time are depicted for the treatment groups in Figure 1. Both ZORVOLEX 18 mg and 35 mg demonstrated efficacy in pain intensity reduction compared with placebo, as measured by the sum of pain intensity difference over 0 to 48 hours after the first dose.
Figure 1. Average Pain Intensity Over 48 Hours for ZORVOLEX 18 mg, ZORVOLEX 35 mg, and Placebo Groups:
Osteoarthritis Pain
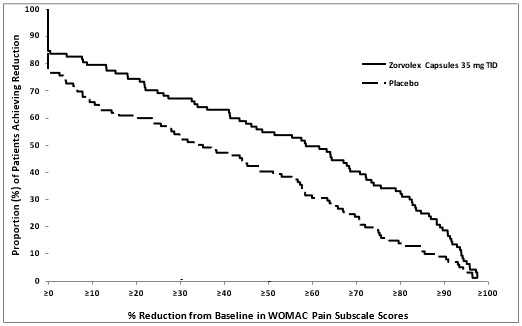
The efficacy of ZORVOLEX in the management of osteoarthritis pain was demonstrated in a single multicenter, randomized, double-blind, placebo-controlled, parallel-arm study comparing ZORVOLEX 35 mg taken twice a day or three times a day and placebo in patients with osteoarthritis of the knee or hip. The study enrolled 305 patients with a mean age of 62 (range 41 to 90 years). Osteoarthritis pain was measured using the Western Ontario and McMaster University Osteoarthritis Index Pain Subscale (WOMAC Pain Subscale). Mean baseline WOMAC Pain Subscale Score across treatment groups was 75 mm using a 0 to 100 mm visual analog scale.
The primary efficacy parameter was the change from baseline at 12 weeks in the WOMAC Pain Subscale. ZORVOLEX 35 mg three times a day reduced osteoarthritis pain compared with placebo, as measured by WOMAC Pain Subscale Score. The distribution (%) of patients achieving various percentage reductions in pain intensity at Week 12 are depicted in Figure 2.
Figure 2. Distribution (%) of Patients Achieving Various Percentage Reductions in Pain Intensity at Week 12:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.