Milsaperidone
Chemical formula: C₂₄H₂₉FN₂O₄ Molecular mass: 428.211 g/mol PubChem compound: 10365268
Mechanism of action
The mechanism of action of milsaperidone in the treatment of schizophrenia in adults and the acute treatment of manic or mixed episodes associated with bipolar I disorder in adults is unknown. However, the efficacy of milsaperidone in these conditions could be mediated through a combination of dopamine type 2 (D2) and serotonin type 2 (5-HT2) antagonism. Milsaperidone and iloperidone rapidly interconvert in vivo. Milsaperidone has an in vitro receptor binding profile similar to iloperidone.
Pharmacodynamic properties
Milsaperidone acts as an antagonist with moderate to strong affinity at the norepinephrine NEα1, NEα2B, NEα2C receptors (Ki values of 8.3, 60, and 16 nM, respectively), the dopamine D1, D2, D3, D4 (Ki values of 96, 16, 68, and 35 nM, respectively), the histamine H1 (Ki value of 26 nM), and the serotonin 5-HT1B, 5-HT2A, and 5-HT2C sites (Ki values of 51, 0.28, and 66 nM, respectively). Milsaperidone has relatively weak affinity at the norepinephrine NEα2A, serotonin 5-HT1A, and 5-HT6 sites (Ki values of 363, 427, and 776 nM, respectively).
Iloperidone acts as an antagonist with high (nM) affinity binding to serotonin 5-HT2A, dopamine D2 and D3 receptors, and norepinephrine NEα1 receptors (Ki values of 5.6, 6.3, 7.1, and 0.36 nM, respectively). Iloperidone has moderate affinity for dopamine D4, and serotonin 5-HT6 and 5-HT7 receptors (Ki values of 25, 43, and 22 nM respectively), and low affinity for the serotonin 5-HT1A, dopamine D1, and histamine H1 receptors (Ki values of 168, 216, and 437 nM, respectively). Iloperidone has no appreciable affinity (Ki>1000 nM) for cholinergic muscarinic receptors. The metabolite P95 only shows affinity for 5-HT2A (Ki value of 3.91) and the NEα1A, NEα1B, NEα1D, and NEα2C receptors (Ki values of 4.7, 2.7, 8.8, and 4.7 nM, respectively).
Cardiac Electrophysiology
In an open-label QTc study in patients with schizophrenia or another psychiatric disorder (n=160), administration of iloperidone (12 mg twice daily) was associated with QTc prolongation of 9 msec. The effect of iloperidone on the QT interval was augmented by the presence of CYP2D6 inhibition (paroxetine 20 mg once daily) or CYP2D6 and CYP3A4 inhibition (paroxetine 20 mg once daily + ketoconazole 200 mg twice daily). Under conditions of metabolic inhibition for both CYP2D6 and CYP3A4, iloperidone 12 mg twice daily was associated with a mean QTcF increase from baseline of about 19 msec.
Pharmacokinetic properties
Following oral administration of iloperidone (iloperidone and milsaperidone rapidly interconvert in vivo), the plasma exposure of milsaperidone increased approximately proportionally over the therapeutic dosage range and plasma exposure of iloperidone increased slightly more than dose proportional. Steady-state concentrations of milsaperidone are attained within 3 to 4 days of dosing. Accumulation of iloperidone is at least 2-fold with twice daily dosing regimen after administration of oral iloperidone tablets.
After administration of oral iloperidone tablets (iloperidone and milsaperidone rapidly interconvert in vivo) in CYP2D6 normal metabolizers, the major metabolite P95, milsaperidone, and iloperidone accounted for approximately 48%, 20% and 9% of the total plasma exposure, respectively. After administration of oral iloperidone tablets in CYP2D6 poor metabolizers, the major metabolite P95, milsaperidone, and iloperidone accounted for 23%, 34%, and 16% of the total exposure, respectively.
Absorption
Following oral administration of milsaperidone or oral iloperidone tablets, no clinically significant differences in the pharmacokinetics of milsaperidone and its metabolites, iloperidone and P95 were observed with the two treatments. Following oral administration of milsaperidone, the time to peak plasma concentrations (Tmax) occurred within 4 hours for milsaperidone, 2 hours for iloperidone, and 6 hours for P95.
Effect of Food
Following administration of milsaperidone with high-fat meal (approximately 1000 calories, 50% fat), no clinically significant differences in the pharmacokinetics of milsaperidone and its metabolites were observed compared to the fasted state.
Distribution
Milsaperidone and iloperidone have an apparent volume of distribution of 1715-2343 L and 1340-2800 L, respectively. At therapeutic concentrations, the unbound fraction in plasma is approximately 8% for milsaperidone, 3% for iloperidone and 8% for P95.
Elimination
In CYP2D6 normal metabolizers, the observed mean elimination half-lives were 26 hours for milsaperidone, 18 hours for iloperidone, and 23 hours for P95. In CYP2D6 poor metabolizers, the mean elimination half-lives were 37 hours for milsaperidone, 33 hours for iloperidone, and 31 hours for P95.
Milsaperidone and iloperidone have an apparent clearance (clearance/bioavailability) of 32 to 69 L/h and 47 to 102 L/h, respectively.
Metabolism
Milsaperidone undergoes oxidation to form iloperidone and iloperidone undergoes a stereospecific carbonyl reduction to form milsaperidone. Elimination of milsaperidone and iloperidone is mainly through hepatic metabolism. Iloperidone is metabolized primarily by 3 biotransformation pathways: carbonyl reduction, hydroxylation (mediated by CYP2D6) and O-demethylation (mediated by CYP3A4).
Excretion
Studies of iloperidone showed the majority of the radioactive materials were recovered in the urine. The mean recovery was 58% in CYP2D6 normal metabolizers and 45% in CYP2D6 poor metabolizers, with feces accounting for 20% in CYP2D6 normal metabolizers and 22% in CYP2D6 poor metabolizers of the administered radioactivity.
Specific Populations
No pharmacokinetics (PK) studies with milsaperidone have been performed in specific populations. The PK of milsaperidone is based on PK studies of iloperidone.
Patients with Renal Impairment
Studies of iloperidone show patients with severe renal impairment (creatinine clearance <30 mL/minute) had minimal effect on Cmax of iloperidone, milsaperidone and P95 compared to those with normal kidney function; AUCinf was increased by 24% for iloperidone, decreased by 6% for milsaperidone and increased by 52% for P95.
Patients with Hepatic Impairment
In patients with moderate hepatic impairment (HI), a 2-fold higher free plasma exposure for milsaperidone was observed and these exposures were variable (these changes are clinically significant), whereas there was a 19% increase in iloperidone exposure and a 5% decrease in P95 exposure. Studies in patients with severe HI have not been conducted. In studies of iloperidone, there were no significant differences in the pharmacokinetics of iloperidone, milsaperidone or P95 (total or unbound) in adult patients with mild HI compared to adults with normal hepatic function.
Drug Interactions Studies
Clinical studies
The effects of fluoxetine and ketoconazole on the exposures of iloperidone, milsaperidone, and P95 are summarized in Figure 1.
Figure 1. Effect of CYP3A4 and CYP2D6 Inhibitors on the Pharmacokinetics of Milsaperidone, Iloperidone, and P95:
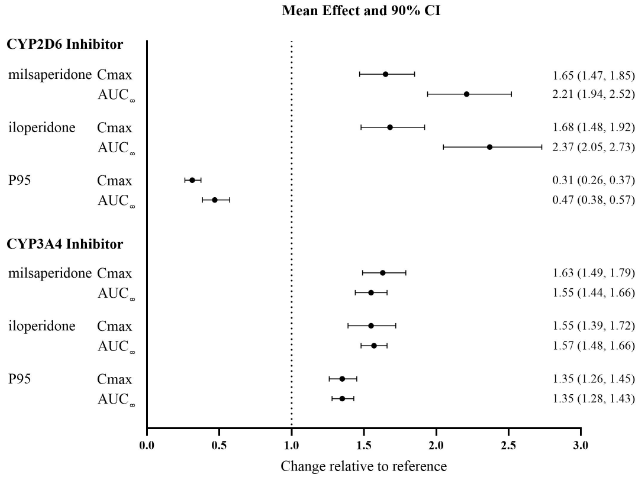
Results are based on single 3 mg doses of iloperidone; top: effect of concomitant administration of a strong CYP2D6 inhibitor (fluoxetine) and bottom: concomitant administration of a strong CYP3A4 inhibitor (ketoconazole). Data are GMRs and 90% CIs. AUC∞: area under the plasma concentration-time curve from time zero extrapolated to infinity; Cmax: maximum plasma concentration, CI: confidence interval; GMRs: geometric mean ratios.
Strong CYP2D6 Inhibitors: Concomitant administration of fluoxetine (a strong CYP2D6 inhibitor) (20 mg twice daily for 21 days) with a single 3 mg dose of iloperidone in CYP2D6 normal metabolizers, increased the AUC of iloperidone and milsaperidone by approximately 2- to 3-fold and decreased the AUC of P95 by one-half. Concomitant administration of a single 3 mg iloperidone dose had no effect on the steady-state pharmacokinetics of fluoxetine.
Concomitant administration of paroxetine (a strong CYP2D6 inhibitor) (20 mg/day for 5-8 days) with multiple doses of iloperidone (8 or 12 mg twice daily) to patients with schizophrenia resulted in increased mean steady-state peak concentrations of iloperidone and milsaperidone, by about 1.6-fold, and decreased mean steady-state peak concentrations of P95 by one-half.
Strong CYP3A4 Inhibitors: Concomitant administration of ketoconazole (a strong CY3A4 inhibitor) (200 mg twice daily for 4 days) with a 3 mg single dose of iloperidone, increased the iloperidone AUC by 57%, the milsaperidone AUC by 55% and the P95 AUC by 35%. Concomitant use of milsaperidone or iloperidone have not been studied with CYP3A4 moderate inhibitors (e.g., erythromycin, grapefruit juice).
Strong CYP2D6 and CYP3A4 Inhibitors: Concomitant administration of paroxetine (20 mg once daily for 10 days) a strong CYP2D6 inhibitor, and ketoconazole (200 mg twice daily), a strong CYP3A4 inhibitor with multiple doses of iloperidone (8 or 12 mg twice daily) in patients with schizophrenia resulted in a 1.4-fold increase in steady-state peak concentrations of both iloperidone and milsaperidone, and a 1.4-fold decrease in steady-state peak concentrations of P95 when compared to paroxetine alone.
Sensitive CYP2D6 Substrates: Concomitant administration of a single 3 mg iloperidone dose with a single 80 mg dextromethorphan dose (a sensitive CYP2D6 substrate) resulted in a 17% increase in AUC and a 26% increase in Cmax for dextromethorphan. Thus, an interaction between iloperidone and other CYP2D6 substrates is unlikely.
Sensitive CYP3A4 substrates: Concomitant administration of midazolam (a sensitive 3A4 substrate) with steady-state iloperidone in patients with schizophrenia showed a less than 50% increase in midazolam AUC and no effect on midazolam Cmax. Thus, an interaction between iloperidone and other CYP3A4 substrates is unlikely.
In Vitro Drug Interaction Studies
Cytochrome P450 (CYP450) Enzymes: Iloperidone is an inhibitor of CYP isozymes 3A4, 3A5 and 2D6. Iloperidone does not inhibit CYP isozymes 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, or 2E1. Iloperidone also does not induce CYP isozymes 1A2, 2C8, 2C9, 2C19, 3A4 and 3A5. Iloperidone is not a substrate of CYP isozymes 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, or 2E1 enzymes. This suggests that an interaction of iloperidone with inhibitors or inducers of these enzymes, or other factors, like smoking, is unlikely.
Transporter Systems: Iloperidone and milsaperidone are not substrates of P-gp and iloperidone is a weak P-gp inhibitor.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.