NEURONTIN Capsule / Film-coated tablet / Solution Ref.[10600] Active ingredients: Gabapentin
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
The precise mechanisms by which gabapentin produces its analgesic and antiepileptic actions are unknown. Gabapentin is structurally related to the neurotransmitter gamma-aminobutyric acid (GABA) but has no effect on GABA binding, uptake, or degradation. In vitro studies have shown that gabapentin binds with high-affinity to the α2δ subunit of voltage-activated calcium channels; however, the relationship of this binding to the therapeutic effects of gabapentin is unknown.
12.3. Pharmacokinetics
All pharmacological actions following gabapentin administration are due to the activity of the parent compound; gabapentin is not appreciably metabolized in humans.
Oral Bioavailability
Gabapentin bioavailability is not dose proportional; i.e., as dose is increased, bioavailability decreases. Bioavailability of gabapentin is approximately 60%, 47%, 34%, 33%, and 27% following 900, 1200, 2400, 3600, and 4800 mg/day given in 3 divided doses, respectively. Food has only a slight effect on the rate and extent of absorption of gabapentin (14% increase in AUC and Cmax).
Distribution
Less than 3% of gabapentin circulates bound to plasma protein. The apparent volume of distribution of gabapentin after 150 mg intravenous administration is 58±6 L (mean ±SD). In patients with epilepsy, steady-state predose (Cmin) concentrations of gabapentin in cerebrospinal fluid were approximately 20% of the corresponding plasma concentrations.
Elimination
Gabapentin is eliminated from the systemic circulation by renal excretion as unchanged drug. Gabapentin is not appreciably metabolized in humans.
Gabapentin elimination half-life is 5 to 7 hours and is unaltered by dose or following multiple dosing. Gabapentin elimination rate constant, plasma clearance, and renal clearance are directly proportional to creatinine clearance. In elderly patients, and in patients with impaired renal function, gabapentin plasma clearance is reduced. Gabapentin can be removed from plasma by hemodialysis.
Specific Populations
Age
The effect of age was studied in subjects 20-80 years of age. Apparent oral clearance (CL/F) of gabapentin decreased as age increased, from about 225 mL/min in those under 30 years of age to about 125 mL/min in those over 70 years of age. Renal clearance (CLr) and CLr adjusted for body surface area also declined with age; however, the decline in the renal clearance of gabapentin with age can largely be explained by the decline in renal function [see Dosage and Administration (2.4) and Use in Specific Populations (8.5)].
Gender
Although no formal study has been conducted to compare the pharmacokinetics of gabapentin in men and women, it appears that the pharmacokinetic parameters for males and females are similar and there are no significant gender differences.
Race
Pharmacokinetic differences due to race have not been studied. Because gabapentin is primarily renally excreted and there are no important racial differences in creatinine clearance, pharmacokinetic differences due to race are not expected.
Pediatric
Gabapentin pharmacokinetics were determined in 48 pediatric subjects between the ages of 1 month and 12 years following a dose of approximately 10 mg/kg. Peak plasma concentrations were similar across the entire age group and occurred 2 to 3 hours postdose. In general, pediatric subjects between 1 month and <5 years of age achieved approximately 30% lower exposure (AUC) than that observed in those 5 years of age and older. Accordingly, oral clearance normalized per body weight was higher in the younger children. Apparent oral clearance of gabapentin was directly proportional to creatinine clearance. Gabapentin elimination half-life averaged 4.7 hours and was similar across the age groups studied.
A population pharmacokinetic analysis was performed in 253 pediatric subjects between 1 month and 13 years of age. Patients received 10 to 65 mg/kg/day given three times a day. Apparent oral clearance (CL/F) was directly proportional to creatinine clearance and this relationship was similar following a single dose and at steady-state. Higher oral clearance values were observed in children <5 years of age compared to those observed in children 5 years of age and older, when normalized per body weight. The clearance was highly variable in infants <1 year of age. The normalized CL/F values observed in pediatric patients 5 years of age and older were consistent with values observed in adults after a single dose. The oral volume of distribution normalized per body weight was constant across the age range.
These pharmacokinetic data indicate that the effective daily dose in pediatric patients with epilepsy ages 3 and 4 years should be 40 mg/kg/day to achieve average plasma concentrations similar to those achieved in patients 5 years of age and older receiving gabapentin at 30 mg/kg/day [see Dosage and Administration (2.2)].
Adult Patients with Renal Impairment
Subjects (N=60) with renal impairment (mean creatinine clearance ranging from 13-114 mL/min) were administered single 400 mg oral doses of gabapentin. The mean gabapentin half-life ranged from about 6.5 hours (patients with creatinine clearance >60 mL/min) to 52 hours (creatinine clearance <30 mL/min) and gabapentin renal clearance from about 90 mL/min (>60 mL/min group) to about 10 mL/min (<30 mL/min). Mean plasma clearance (CL/F) decreased from approximately 190 mL/min to 20 mL/min [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)]. Pediatric patients with renal insufficiency have not been studied.
Hemodialysis
In a study in anuric adult subjects (N=11), the apparent elimination half-life of gabapentin on nondialysis days was about 132 hours; during dialysis the apparent half-life of gabapentin was reduced to 3.8 hours. Hemodialysis thus has a significant effect on gabapentin elimination in anuric subjects [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)].
Hepatic Disease
Because gabapentin is not metabolized, no study was performed in patients with hepatic impairment.
Drug Interactions
In Vitro Studies
In vitro studies were conducted to investigate the potential of gabapentin to inhibit the major cytochrome P450 enzymes (CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4) that mediate drug and xenobiotic metabolism using isoform selective marker substrates and human liver microsomal preparations. Only at the highest concentration tested (171 mcg/mL; 1 mM) was a slight degree of inhibition (14% to 30%) of isoform CYP2A6 observed. No inhibition of any of the other isoforms tested was observed at gabapentin concentrations up to 171 mcg/mL (approximately 15 times the Cmax at 3600 mg/day).
In Vivo Studies
The drug interaction data described in this section were obtained from studies involving healthy adults and adult patients with epilepsy.
Phenytoin: In a single (400 mg) and multiple dose (400 mg three times a day) study of NEURONTIN in epileptic patients (N=8) maintained on phenytoin monotherapy for at least 2 months, gabapentin had no effect on the steady-state trough plasma concentrations of phenytoin and phenytoin had no effect on gabapentin pharmacokinetics.
Carbamazepine: Steady-state trough plasma carbamazepine and carbamazepine 10, 11 epoxide concentrations were not affected by concomitant gabapentin (400 mg three times a day; N=12) administration. Likewise, gabapentin pharmacokinetics were unaltered by carbamazepine administration.
Valproic Acid: The mean steady-state trough serum valproic acid concentrations prior to and during concomitant gabapentin administration (400 mg three times a day; N=17) were not different and neither were gabapentin pharmacokinetic parameters affected by valproic acid.
Phenobarbital: Estimates of steady-state pharmacokinetic parameters for phenobarbital or gabapentin (300 mg three times a day; N=12) are identical whether the drugs are administered alone or together.
Naproxen: Coadministration (N=18) of naproxen sodium capsules (250 mg) with NEURONTIN (125 mg) appears to increase the amount of gabapentin absorbed by 12% to 15%. Gabapentin had no effect on naproxen pharmacokinetic parameters. These doses are lower than the therapeutic doses for both drugs. The magnitude of interaction within the recommended dose ranges of either drug is not known.
Hydrocodone: Coadministration of NEURONTIN (125 to 500 mg; N=48) decreases hydrocodone (10 mg; N=50) Cmax and AUC values in a dose-dependent manner relative to administration of hydrocodone alone; Cmax and AUC values are 3% to 4% lower, respectively, after administration of 125 mg NEURONTIN and 21% to 22% lower, respectively, after administration of 500 mg NEURONTIN. The mechanism for this interaction is unknown. Hydrocodone increases gabapentin AUC values by 14%. The magnitude of interaction at other doses is not known.
Morphine: A literature article reported that when a 60 mg controlled-release morphine capsule was administered 2 hours prior to a 600 mg NEURONTIN capsule (N=12), mean gabapentin AUC increased by 44% compared to gabapentin administered without morphine. Morphine pharmacokinetic parameter values were not affected by administration of NEURONTIN 2 hours after morphine. The magnitude of interaction at other doses is not known.
Cimetidine: In the presence of cimetidine at 300 mg four times a day (N=12), the mean apparent oral clearance of gabapentin fell by 14% and creatinine clearance fell by 10%. Thus, cimetidine appeared to alter the renal excretion of both gabapentin and creatinine, an endogenous marker of renal function. This small decrease in excretion of gabapentin by cimetidine is not expected to be of clinical importance. The effect of gabapentin on cimetidine was not evaluated.
Oral Contraceptive: Based on AUC and half-life, multiple-dose pharmacokinetic profiles of norethindrone and ethinyl estradiol following administration of tablets containing 2.5 mg of norethindrone acetate and 50 mcg of ethinyl estradiol were similar with and without coadministration of gabapentin (400 mg three times a day; N=13). The Cmax of norethindrone was 13% higher when it was coadministered with gabapentin; this interaction is not expected to be of clinical importance.
Antacid (Maalox) (aluminum hydroxide, magnesium hydroxide): Antacid (Maalox) containing magnesium and aluminum hydroxides reduced the mean bioavailability of gabapentin (N=16) by about 20%. This decrease in bioavailability was about 10% when gabapentin was administered 2 hours after Maalox.
Probenecid: Probenecid is a blocker of renal tubular secretion. Gabapentin pharmacokinetic parameters without and with probenecid were comparable. This indicates that gabapentin does not undergo renal tubular secretion by the pathway that is blocked by probenecid.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Gabapentin was administered orally to mice and rats in 2-year carcinogenicity studies. No evidence of drug-related carcinogenicity was observed in mice treated at doses up to 2000 mg/kg/day. At 2000 mg/kg, the plasma gabapentin exposure (AUC) in mice was approximately 2 times that in humans at the MRHD of 3600 mg/day. In rats, increases in the incidence of pancreatic acinar cell adenoma and carcinoma were found in male rats receiving the highest dose (2000 mg/kg), but not at doses of 250 or 1000 mg/kg/day. At 1000 mg/kg, the plasma gabapentin exposure (AUC) in rats was approximately 5 times that in humans at the MRHD.
Studies designed to investigate the mechanism of gabapentin-induced pancreatic carcinogenesis in rats indicate that gabapentin stimulates DNA synthesis in rat pancreatic acinar cells in vitro and, thus, may be acting as a tumor promoter by enhancing mitogenic activity. It is not known whether gabapentin has the ability to increase cell proliferation in other cell types or in other species, including humans.
Mutagenesis
Gabapentin did not demonstrate mutagenic or genotoxic potential in in vitro (Ames test, HGPRT forward mutation assay in Chinese hamster lung cells) and in vivo (chromosomal aberration and micronucleus test in Chinese hamster bone marrow, mouse micronucleus, unscheduled DNA synthesis in rat hepatocytes) assays.
Impairment of Fertility
No adverse effects on fertility or reproduction were observed in rats at doses up to 2000 mg/kg. At 2000 mg/kg, the plasma gabapentin exposure (AUC) in rats is approximately 8 times that in humans at the MRHD.
14. Clinical Studies
14.1 Postherpetic Neuralgia
NEURONTIN was evaluated for the management of postherpetic neuralgia (PHN) in two randomized, double-blind, placebo-controlled, multicenter studies. The intent-to-treat (ITT) population consisted of a total of 563 patients with pain for more than 3 months after healing of the herpes zoster skin rash (Table 6).
Table 6. Controlled PHN Studies: Duration, Dosages, and Number of Patients:
| Study | Study Duration | Gabapentin (mg/day)* Target Dose | Patients Receiving Gabapentin | Patients Receiving Placebo |
|---|---|---|---|---|
| 1 | 8 weeks | 3600 | 113 | 116 |
| 2 | 7 weeks | 1800, 2400 | 223 | 111 |
| Total | 336 | 227 | ||
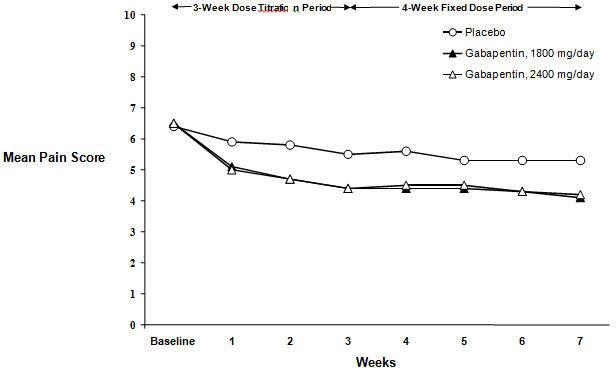
Each study included a 7- or 8-week double-blind phase (3 or 4 weeks of titration and 4 weeks of fixed dose). Patients initiated treatment with titration to a maximum of 900 mg/day gabapentin over 3 days. Dosages were then to be titrated in 600 to 1200 mg/day increments at 3- to 7-day intervals to the target dose over 3 to 4 weeks. Patients recorded their pain in a daily diary using an 11-point numeric pain rating scale ranging from 0 (no pain) to 10 (worst possible pain). A mean pain score during baseline of at least 4 was required for randomization. Analyses were conducted using the ITT population (all randomized patients who received at least one dose of study medication).
Both studies demonstrated efficacy compared to placebo at all doses tested.
The reduction in weekly mean pain scores was seen by Week 1 in both studies, and were maintained to the end of treatment. Comparable treatment effects were observed in all active treatment arms. Pharmacokinetic/pharmacodynamic modeling provided confirmatory evidence of efficacy across all doses. Figures 1 and 2 show pain intensity scores over time for Studies 1 and 2.
Figure 1. Weekly Mean Pain Scores (Observed Cases in ITT Population): Study 1:
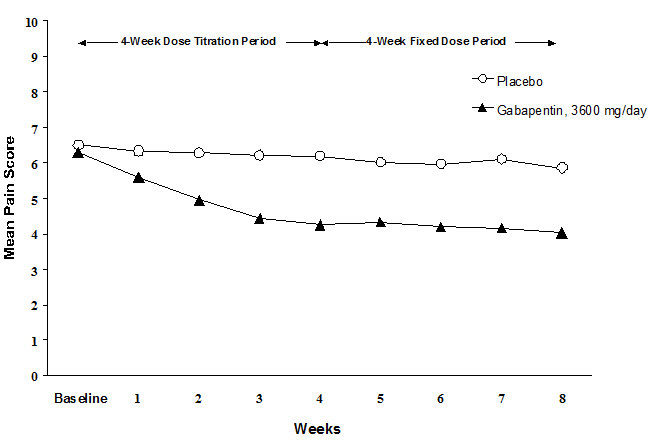
Figure 2. Weekly Mean Pain Scores (Observed Cases in ITT Population): Study 2:
The proportion of responders (those patients reporting at least 50% improvement in endpoint pain score compared to baseline) was calculated for each study (Figure 3).
Figure 3. Proportion of Responders (patients with ≥50% reduction in pain score) at Endpoint: Controlled PHN Studies:
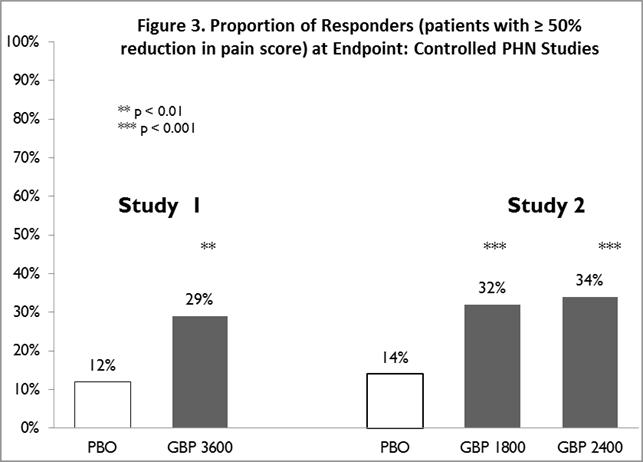
14.2 Epilepsy for Partial Onset Seizures (Adjunctive Therapy)
The effectiveness of NEURONTIN as adjunctive therapy (added to other antiepileptic drugs) was established in multicenter placebo-controlled, double-blind, parallel-group clinical trials in adult and pediatric patients (3 years and older) with refractory partial seizures.
Evidence of effectiveness was obtained in three trials conducted in 705 patients (age 12 years and above) and one trial conducted in 247 pediatric patients (3 to 12 years of age). The patients enrolled had a history of at least 4 partial seizures per month in spite of receiving one or more antiepileptic drugs at therapeutic levels and were observed on their established antiepileptic drug regimen during a 12-week baseline period (6 weeks in the study of pediatric patients). In patients continuing to have at least 2 (or 4 in some studies) seizures per month, NEURONTIN or placebo was then added on to the existing therapy during a 12-week treatment period. Effectiveness was assessed primarily on the basis of the percent of patients with a 50% or greater reduction in seizure frequency from baseline to treatment (the “responder rate”) and a derived measure called response ratio, a measure of change defined as (T – B)/(T + B), in which B is the patient’s baseline seizure frequency and T is the patient’s seizure frequency during treatment. Response ratio is distributed within the range -1 to +1. A zero value indicates no change while complete elimination of seizures would give a value of -1; increased seizure rates would give positive values. A response ratio of -0.33 corresponds to a 50% reduction in seizure frequency. The results given below are for all partial seizures in the intent-to-treat (all patients who received any doses of treatment) population in each study, unless otherwise indicated.
One study compared NEURONTIN 1200 mg/day, in three divided doses with placebo. Responder rate was 23% (14/61) in the NEURONTIN group and 9% (6/66) in the placebo group; the difference between groups was statistically significant. Response ratio was also better in the NEURONTIN group (-0.199) than in the placebo group (-0.044), a difference that also achieved statistical significance.
A second study compared primarily NEURONTIN 1200 mg/day, in three divided doses (N=101), with placebo (N=98). Additional smaller NEURONTIN dosage groups (600 mg/day, N=53; 1800 mg/day, N=54) were also studied for information regarding dose response. Responder rate was higher in the NEURONTIN 1200 mg/day group (16%) than in the placebo group (8%), but the difference was not statistically significant. The responder rate at 600 mg (17%) was also not significantly higher than in the placebo, but the responder rate in the 1800 mg group (26%) was statistically significantly superior to the placebo rate. Response ratio was better in the NEURONTIN 1200 mg/day group (-0.103) than in the placebo group (-0.022); but this difference was also not statistically significant (p = 0.224). A better response was seen in the NEURONTIN 600 mg/day group (-0.105) and 1800 mg/day group (-0.222) than in the 1200 mg/day group, with the 1800 mg/day group achieving statistical significance compared to the placebo group.
A third study compared NEURONTIN 900 mg/day, in three divided doses (N=111), and placebo (N=109). An additional NEURONTIN 1200 mg/day dosage group (N=52) provided dose-response data. A statistically significant difference in responder rate was seen in the NEURONTIN 900 mg/day group (22%) compared to that in the placebo group (10%). Response ratio was also statistically significantly superior in the NEURONTIN 900 mg/day group (-0.119) compared to that in the placebo group (-0.027), as was response ratio in 1200 mg/day NEURONTIN (-0.184) compared to placebo.
Analyses were also performed in each study to examine the effect of NEURONTIN on preventing secondarily generalized tonic-clonic seizures. Patients who experienced a secondarily generalized tonic-clonic seizure in either the baseline or in the treatment period in all three placebo-controlled studies were included in these analyses. There were several response ratio comparisons that showed a statistically significant advantage for NEURONTIN compared to placebo and favorable trends for almost all comparisons.
Analysis of responder rate using combined data from all three studies and all doses (N=162, NEURONTIN; N=89, placebo) also showed a significant advantage for NEURONTIN over placebo in reducing the frequency of secondarily generalized tonic-clonic seizures.
In two of the three controlled studies, more than one dose of NEURONTIN was used. Within each study, the results did not show a consistently increased response to dose. However, looking across studies, a trend toward increasing efficacy with increasing dose is evident (see Figure 4).
Figure 4. Responder Rate in Patients Receiving NEURONTIN Expressed as a Difference from Placebo by Dose and Study: Adjunctive Therapy Studies in Patients ≥12 Years of Age with Partial Seizures:
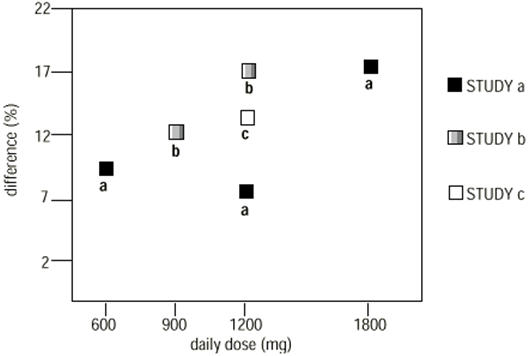
In the figure, treatment effect magnitude, measured on the Y axis in terms of the difference in the proportion of gabapentin and placebo-assigned patients attaining a 50% or greater reduction in seizure frequency from baseline, is plotted against the daily dose of gabapentin administered (X axis).
Although no formal analysis by gender has been performed, estimates of response (Response Ratio) derived from clinical trials (398 men, 307 women) indicate no important gender differences exist. There was no consistent pattern indicating that age had any effect on the response to NEURONTIN. There were insufficient numbers of patients of races other than Caucasian to permit a comparison of efficacy among racial groups.
A fourth study in pediatric patients age 3 to 12 years compared 25-35 mg/kg/day NEURONTIN (N=118) with placebo (N=127). For all partial seizures in the intent-to-treat population, the response ratio was statistically significantly better for the NEURONTIN group (-0.146) than for the placebo group (-0.079). For the same population, the responder rate for NEURONTIN (21%) was not significantly different from placebo (18%).
A study in pediatric patients age 1 month to 3 years compared 40 mg/kg/day NEURONTIN (N=38) with placebo (N=38) in patients who were receiving at least one marketed antiepileptic drug and had at least one partial seizure during the screening period (within 2 weeks prior to baseline). Patients had up to 48 hours of baseline and up to 72 hours of double-blind video EEG monitoring to record and count the occurrence of seizures. There were no statistically significant differences between treatments in either the response ratio or responder rate.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.