BYSANTI Tablet Ref.[116258] Active ingredients: Milsaperidone
Source: FDA, National Drug Code (US) Revision Year: 2026
12.1. Mechanism of Action
The mechanism of action of milsaperidone in the treatment of schizophrenia in adults and the acute treatment of manic or mixed episodes associated with bipolar I disorder in adults is unknown. However, the efficacy of milsaperidone in these conditions could be mediated through a combination of dopamine type 2 (D2) and serotonin type 2 (5-HT2) antagonism. Milsaperidone and iloperidone rapidly interconvert in vivo. Milsaperidone has an in vitro receptor binding profile similar to iloperidone.
12.2. Pharmacodynamics
Milsaperidone acts as an antagonist with moderate to strong affinity at the norepinephrine NEα1, NEα2B, NEα2C receptors (Ki values of 8.3, 60, and 16 nM, respectively), the dopamine D1, D2, D3, D4 (Ki values of 96, 16, 68, and 35 nM, respectively), the histamine H1 (Ki value of 26 nM), and the serotonin 5-HT1B, 5-HT2A, and 5-HT2C sites (Ki values of 51, 0.28, and 66 nM, respectively). Milsaperidone has relatively weak affinity at the norepinephrine NEα2A, serotonin 5-HT1A, and 5-HT6 sites (Ki values of 363, 427, and 776 nM, respectively).
Iloperidone acts as an antagonist with high (nM) affinity binding to serotonin 5-HT2A, dopamine D2 and D3 receptors, and norepinephrine NEα1 receptors (Ki values of 5.6, 6.3, 7.1, and 0.36 nM, respectively). Iloperidone has moderate affinity for dopamine D4, and serotonin 5-HT6 and 5-HT7 receptors (Ki values of 25, 43, and 22 nM respectively), and low affinity for the serotonin 5-HT1A, dopamine D1, and histamine H1 receptors (Ki values of 168, 216, and 437 nM, respectively). Iloperidone has no appreciable affinity (Ki>1000 nM) for cholinergic muscarinic receptors. The metabolite P95 only shows affinity for 5-HT2A (Ki value of 3.91) and the NEα1A, NEα1B, NEα1D, and NEα2C receptors (Ki values of 4.7, 2.7, 8.8, and 4.7 nM, respectively).
Cardiac Electrophysiology
In an open-label QTc study in patients with schizophrenia or another psychiatric disorder (n=160), administration of iloperidone (12 mg twice daily) was associated with QTc prolongation of 9 msec. The effect of iloperidone on the QT interval was augmented by the presence of CYP2D6 inhibition (paroxetine 20 mg once daily) or CYP2D6 and CYP3A4 inhibition (paroxetine 20 mg once daily + ketoconazole 200 mg twice daily). Under conditions of metabolic inhibition for both CYP2D6 and CYP3A4, iloperidone 12 mg twice daily was associated with a mean QTcF increase from baseline of about 19 msec [see Warnings and Precautions (5.3)].
12.3. Pharmacokinetics
Following oral administration of iloperidone (iloperidone and milsaperidone rapidly interconvert in vivo), the plasma exposure of milsaperidone increased approximately proportionally over the therapeutic dosage range and plasma exposure of iloperidone increased slightly more than dose proportional. Steady-state concentrations of milsaperidone are attained within 3 to 4 days of dosing. Accumulation of iloperidone is at least 2-fold with twice daily dosing regimen after administration of oral iloperidone tablets.
After administration of oral iloperidone tablets (iloperidone and milsaperidone rapidly interconvert in vivo) in CYP2D6 normal metabolizers, the major metabolite P95, milsaperidone, and iloperidone accounted for approximately 48%, 20% and 9% of the total plasma exposure, respectively. After administration of oral iloperidone tablets in CYP2D6 poor metabolizers, the major metabolite P95, milsaperidone, and iloperidone accounted for 23%, 34%, and 16% of the total exposure, respectively.
Absorption
Following oral administration of BYSANTI or oral iloperidone tablets, no clinically significant differences in the pharmacokinetics of milsaperidone and its metabolites, iloperidone and P95 were observed with the two treatments. Following oral administration of BYSANTI, the time to peak plasma concentrations (Tmax) occurred within 4 hours for milsaperidone, 2 hours for iloperidone, and 6 hours for P95.
Effect of Food
Following administration of BYSANTI with high-fat meal (approximately 1000 calories, 50% fat), no clinically significant differences in the pharmacokinetics of milsaperidone and its metabolites were observed compared to the fasted state.
Distribution
Milsaperidone and iloperidone have an apparent volume of distribution of 1715-2343 L and 1340-2800 L, respectively. At therapeutic concentrations, the unbound fraction in plasma is approximately 8% for milsaperidone, 3% for iloperidone and 8% for P95.
Elimination
In CYP2D6 normal metabolizers, the observed mean elimination half-lives were 26 hours for milsaperidone, 18 hours for iloperidone, and 23 hours for P95. In CYP2D6 poor metabolizers, the mean elimination half-lives were 37 hours for milsaperidone, 33 hours for iloperidone, and 31 hours for P95.
Milsaperidone and iloperidone have an apparent clearance (clearance/bioavailability) of 32 to 69 L/h and 47 to 102 L/h, respectively.
Metabolism
Milsaperidone undergoes oxidation to form iloperidone and iloperidone undergoes a stereospecific carbonyl reduction to form milsaperidone. Elimination of milsaperidone and iloperidone is mainly through hepatic metabolism. Iloperidone is metabolized primarily by 3 biotransformation pathways: carbonyl reduction, hydroxylation (mediated by CYP2D6) and O-demethylation (mediated by CYP3A4).
Excretion
Studies of iloperidone showed the majority of the radioactive materials were recovered in the urine. The mean recovery was 58% in CYP2D6 normal metabolizers and 45% in CYP2D6 poor metabolizers, with feces accounting for 20% in CYP2D6 normal metabolizers and 22% in CYP2D6 poor metabolizers of the administered radioactivity.
Specific Populations
No pharmacokinetics (PK) studies with BYSANTI have been performed in specific populations. The PK of milsaperidone is based on PK studies of iloperidone.
Patients with Renal Impairment
Studies of iloperidone show patients with severe renal impairment (creatinine clearance <30 mL/minute) had minimal effect on Cmax of iloperidone, milsaperidone and P95 compared to those with normal kidney function; AUCinf was increased by 24% for iloperidone, decreased by 6% for milsaperidone and increased by 52% for P95.
Patients with Hepatic Impairment
In patients with moderate hepatic impairment (HI), a 2-fold higher free plasma exposure for milsaperidone was observed and these exposures were variable (these changes are clinically significant), whereas there was a 19% increase in iloperidone exposure and a 5% decrease in P95 exposure. Studies in patients with severe HI have not been conducted. In studies of iloperidone, there were no significant differences in the pharmacokinetics of iloperidone, milsaperidone or P95 (total or unbound) in adult patients with mild HI compared to adults with normal hepatic function. [see Dosage and Administration (2.3), Use in Specific Populations (8.6)].
Drug Interactions Studies
Clinical studies
The effects of fluoxetine and ketoconazole on the exposures of iloperidone, milsaperidone, and P95 are summarized in Figure 1.
Figure 1. Effect of CYP3A4 and CYP2D6 Inhibitors on the Pharmacokinetics of Milsaperidone, Iloperidone, and P95:
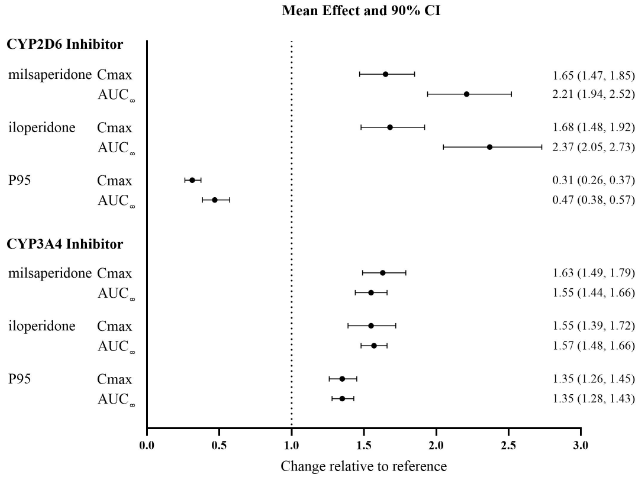
Results are based on single 3 mg doses of iloperidone; top: effect of concomitant administration of a strong CYP2D6 inhibitor (fluoxetine) and bottom: concomitant administration of a strong CYP3A4 inhibitor (ketoconazole). Data are GMRs and 90% CIs. AUC∞: area under the plasma concentration-time curve from time zero extrapolated to infinity; Cmax: maximum plasma concentration, CI: confidence interval; GMRs: geometric mean ratios.
Strong CYP2D6 Inhibitors: Concomitant administration of fluoxetine (a strong CYP2D6 inhibitor) (20 mg twice daily for 21 days) with a single 3 mg dose of iloperidone in CYP2D6 normal metabolizers, increased the AUC of iloperidone and milsaperidone by approximately 2- to 3-fold and decreased the AUC of P95 by one-half [see Drug Interactions (7.1)]. Concomitant administration of a single 3 mg iloperidone dose had no effect on the steady-state pharmacokinetics of fluoxetine.
Concomitant administration of paroxetine (a strong CYP2D6 inhibitor) (20 mg/day for 5-8 days) with multiple doses of iloperidone (8 or 12 mg twice daily) to patients with schizophrenia resulted in increased mean steady-state peak concentrations of iloperidone and milsaperidone, by about 1.6-fold, and decreased mean steady-state peak concentrations of P95 by one-half [see Drug Interactions (7.1)].
Strong CYP3A4 Inhibitors: Concomitant administration of ketoconazole (a strong CY3A4 inhibitor) (200 mg twice daily for 4 days) with a 3 mg single dose of iloperidone, increased the iloperidone AUC by 57%, the milsaperidone AUC by 55% and the P95 AUC by 35%. Concomitant use of BYSANTI or iloperidone have not been studied with CYP3A4 moderate inhibitors (e.g., erythromycin, grapefruit juice).
Strong CYP2D6 and CYP3A4 Inhibitors: Concomitant administration of paroxetine (20 mg once daily for 10 days) a strong CYP2D6 inhibitor, and ketoconazole (200 mg twice daily), a strong CYP3A4 inhibitor with multiple doses of iloperidone (8 or 12 mg twice daily) in patients with schizophrenia resulted in a 1.4-fold increase in steady-state peak concentrations of both iloperidone and milsaperidone, and a 1.4-fold decrease in steady-state peak concentrations of P95 when compared to paroxetine alone [see Drug Interactions (7.1)].
Sensitive CYP2D6 Substrates: Concomitant administration of a single 3 mg iloperidone dose with a single 80 mg dextromethorphan dose (a sensitive CYP2D6 substrate) resulted in a 17% increase in AUC and a 26% increase in Cmax for dextromethorphan. Thus, an interaction between iloperidone and other CYP2D6 substrates is unlikely.
Sensitive CYP3A4 substrates: Concomitant administration of midazolam (a sensitive 3A4 substrate) with steady-state iloperidone in patients with schizophrenia showed a less than 50% increase in midazolam AUC and no effect on midazolam Cmax. Thus, an interaction between iloperidone and other CYP3A4 substrates is unlikely.
In Vitro Drug Interaction Studies
Cytochrome P450 (CYP450) Enzymes: Iloperidone is an inhibitor of CYP isozymes 3A4, 3A5 and 2D6. Iloperidone does not inhibit CYP isozymes 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, or 2E1. Iloperidone also does not induce CYP isozymes 1A2, 2C8, 2C9, 2C19, 3A4 and 3A5. Iloperidone is not a substrate of CYP isozymes 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, or 2E1 enzymes. This suggests that an interaction of iloperidone with inhibitors or inducers of these enzymes, or other factors, like smoking, is unlikely.
Transporter Systems: Iloperidone and milsaperidone are not substrates of P-gp and iloperidone is a weak P-gp inhibitor.
12.5. Pharmacogenomics
Milsaperidone and iloperidone (iloperidone and milsaperidone rapidly interconvert in vivo) are metabolized by CYP2D6 [see Clinical Pharmacology (12.3)]. The gene encoding CYP2D6 has variants that affect CYP2D6 metabolic function. CYP2D6 poor metabolizers are individuals with two nonfunctional alleles (e.g., *3/*4, *4/*4, *5/*5, *5/*6), and as a result they have no CYP2D6 enzyme activity.
Pharmacokinetic data from CYP2D6 poor metabolizers (n=8) treated with iloperidone demonstrated milsaperidone AUC was 85% higher, iloperidone AUC was 47% higher, and the P95 metabolite AUC was 85% lower compared to the AUC in CYP2D6 normal metabolizers (n=18) [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
The pharmacokinetics of milsaperidone, iloperidone, and P95 were not fully evaluated in CYP2D6 ultrarapid and intermediate metabolizers.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime carcinogenicity studies were conducted with iloperidone (iloperidone and milsaperidone rapidly interconvert in vivo) in CD-1 mice and Sprague Dawley rats. Iloperidone was administered orally at doses of 2.5, 5, and 10 mg/kg/day to CD-1 mice and 4, 8, and 16 mg/kg/day to Sprague Dawley rats (0.5, 1, and 2 times and 1.6, 3.2, and 6.5 times, respectively, the MRHD of 24 mg/day on a mg/m² basis). The safety margins for milsaperidone are expected to be the same as those seen with iloperidone because exposure to iloperidone, milsaperidone, and their major metabolites are similar when either iloperidone tablets or BYSANTI (milsaperidone) tablets are administered in humans. There was an increased incidence of malignant mammary gland tumors in female mice treated with the lowest dose (2.5 mg/kg/day) only. There were no treatment-related increases in neoplasia in rats.
The carcinogenic potential of the milsaperidone metabolite P95, which is a major circulating metabolite of milsaperidone in humans but is not present at significant amounts in mice or rats, was assessed in a lifetime carcinogenicity study in Wistar rats at oral doses of 25, 75, and 200 mg/kg/day in males and 50, 150, and 250 (reduced from 400) mg/kg/day in females. Drug-related neoplastic changes occurred in males, in the pituitary gland (pars distalis adenoma) at all doses and in the pancreas (islet cell adenoma) at the high dose. Plasma levels of P95 (AUC) in males at the tested doses (25, 75, and 200 mg/kg/day) were approximately 0.4, 3, and 23 times, respectively, the human exposure to P95 at the MRHD of milsaperidone.
Mutagenesis
Milsaperidone was not mutagenic in the Bacterial Reverse Mutation (Ames) Test conducted in multiple bacterial strains with and without metabolic activation. Milsaperidone did not induce structural chromosomal damage in an in vitro cytogenetic assay using Chinese Hamster Ovary (CHO) cells with and without metabolic activation.
Iloperidone was negative in the Ames test and in the in vivo mouse bone marrow and rat liver micronucleus tests. Iloperidone induced chromosomal aberrations in Chinese Hamster Ovary (CHO) cells in vitro at concentrations which also caused some cytotoxicity.
The iloperidone metabolite P95 was negative in the Ames test, the V79 chromosome aberration test, and an in vivo mouse bone marrow micronucleus test.
Impairment of Fertility
Iloperidone decreased fertility at 12 and 36 mg/kg in a study in which both male and female rats were treated. The no-effect dose was 4 mg/kg, which is 1.6 times the MRHD of 24 mg/day on a mg/m² basis.
14. Clinical Studies
14.1 Schizophrenia
The effectiveness of BYSANTI in the treatment of schizophrenia in adults has been established from adequate and well- controlled studies of iloperidone tablets (referred to as "iloperidone" (iloperidone and milsaperidone rapidly interconvert in vivo)) in adults with schizophrenia. Below is a display of the efficacy results of iloperidone in these adequate and well-controlled studies.
Iloperidone was studied in two placebo- and active-controlled short-term trials (a 6-week trial (Study 1) and a 4-week trial (Study 2)) and one long-term placebo-controlled randomized withdrawal trial (Study 3) in adult patients who met the DSM-III/IV criteria for schizophrenia.
In Studies 1, 2, and 3, three instruments were used to assess psychiatric signs and symptoms: the Positive and Negative Syndrome Scale (PANSS), the Brief Psychiatric Rating Scale (BPRS) (both multi-item inventories of general psychopathology), and the Clinical Global Impression (CGI) assessment which reflects the impression of a skilled observer, fully familiar with the manifestations of schizophrenia, about the overall clinical state of the patient.
Study 1
Study 1 (n=706) included two flexible dosage ranges of iloperidone (12 mg to 16 mg/day or 20 mg to 24 mg/day) compared to placebo and an active control (risperidone).
- For the 12 mg to 16 mg/day group, the iloperidone titration schedule was 1 mg twice daily on Days 1 and 2, 2 mg twice daily on Days 3 and 4, 4 mg twice daily on Days 5 and 6, and 6 mg twice daily on Day 7.
- For the 20 mg to 24 mg/day group, the iloperidone titration schedule was 1 mg twice daily on Day 1, 2 mg twice daily on Day 2, 4 mg twice daily on Day 3, 6 mg twice daily on Days 4 and 5, 8 mg twice daily on Day 6, and 10 mg twice daily on Day 7.
In Study 1, the primary endpoint was change from baseline on the BPRS total score at the end of treatment (Day 42). Both the 12 mg to 16 mg/day and the 20 mg to 24 mg/day iloperidone treatment groups were superior to the placebo group on the BPRS total score. The active control antipsychotic drug (risperidone) appeared to be superior to iloperidone in this trial within the first 2 weeks, a finding that may in part be explained by the more rapid titration that was possible for risperidone. For patients in Study 1 who remained on treatment for at least 2 weeks, iloperidone appeared to have had comparable efficacy to risperidone.
Study 2
Study 2 (NCT00254202) (n=604) compared one fixed-dose of iloperidone (24 mg/day) to placebo and an active control (ziprasidone).
- The titration schedule for Study 2 was similar to the titration schedule for Study 1. In Study 2, the titration of iloperidone started at 1 mg twice daily on Day 1 and increased to 2, 4, 6, 8, 10, and 12 mg twice daily on Days 2, 3, 4, 5, 6, and 7, respectively.
In Study 2, the primary endpoint was change from baseline on the PANSS total score at the end of treatment (Day 28). The 24 mg/day iloperidone treatment group was superior to the placebo treatment group in the PANSS total score. In Study 2, iloperidone appeared to have similar efficacy as ziprasidone which also needed a slow titration to the target dosage.
Study 3
Study 3 (NCT01291511) included clinically stable adult outpatients (n=303) who met DSM-IV criteria for schizophrenia. After a one-week iloperidone titration, patients who remained clinically stable received 12 weeks of open-label treatment with a flexible iloperidone dosage (4 mg to 12 mg administered twice daily (8 mg to 24 mg per day, respectively). Stabilization during the open-label phase was defined as being on an established iloperidone dosage that was unchanged due to efficacy in the 4 weeks prior to randomization, having CGI-Severity score of ≤4 and PANSS total score ≤70, a score of ≤4 on each of the following individual PANSS items (P1-delusions, P2-conceptual disorganization, P3-hallucinatory behavior, P6-suspiciousness/persecution, P7-hostility, or G8-uncooperativeness), and no hospitalization or increase in level of care to treat exacerbations.
After the open-label phase in Study 3, patients were randomized to receive placebo or to continue on their current iloperidone dosage (4 to 12 mg twice daily (8 mg to 24 mg/day, respectively)) and were observed for possible relapse during the double-blind phase. Relapse or impending relapse during the double-blind phase was defined as any of the following: hospitalization due to worsening of schizophrenia, increase (worsening) of the PANSS total score ≥30%, CGI-Improvement score ≥6, or the patient had suicidal, homicidal, or aggressive behavior, or needed any other antipsychotic drug.
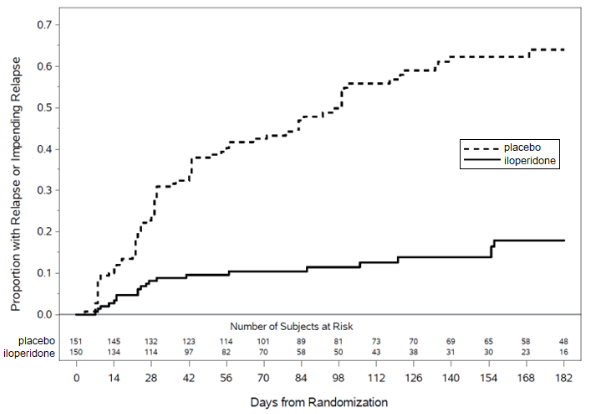
Based on the interim analysis in Study 3, an independent data monitoring committee decided the study should be discontinued early due to evidence of efficacy. Based on results from the interim analysis, which were confirmed by the final analysis dataset, patients treated with iloperidone experienced a statistically significant longer time to relapse or impending relapse than patients who received placebo. Figure 2 displays the estimated cumulative proportion of patients with relapse or impending relapse in Study 3.
Figure 2. Kaplan Meier Estimation of Percent Relapse/Impending Relapse in Adult Patients with Schizophrenia (Study 3):
14.2 Manic or Mixed Episodes Associated with Bipolar I Disorder
The effectiveness of BYSANTI in the acute treatment of manic or mixed episodes associated with bipolar I disorder has been established from an adequate and well-controlled study of iloperidone tablets (referred to as "iloperidone" (iloperidone and milsaperidone rapidly interconvert in vivo)) in adults with manic or mixed episodes associated with bipolar I disorder. Below is a display of the efficacy results of iloperidone in this adequate and well-controlled study.
Iloperidone was studied in one multicenter, randomized, double-blind, placebo-controlled, 4-week study (n=392) that enrolled adult patients who met the DSM-5 criteria for bipolar I disorder, manic or mixed type (Study 4; NCT04819776). In Study 4, the demographic and baseline characteristics were similar in the iloperidone and placebo groups. The median age was 46 (range 18 to 65); 45% were female; 64% were White, and 28% were Black or African American.
In Study 4, manic symptoms were assessed with the Young Mania Rating Scale (YMRS). The YMRS is an 11-item clinician rated scale traditionally used to assess the degree of manic symptomatology. YMRS total scores may range from 0 to 60 with a higher score reflecting greater severity.
In Study 4, patients received one fixed-dose of iloperidone (12 mg twice daily (24 mg/day)) after a four-day titration (1 mg twice daily on Day 1, 3 mg twice daily on Day 2, 6 mg twice daily on Day 3, 9 mg twice daily on Day 4, and then 12 mg twice daily on Day 5) or placebo. CYP2D6 poor metabolizers who received iloperidone in the study received a fixed-dose of iloperidone (6 mg twice daily (12 mg/day)) after a two-day titration (i.e., 1 mg twice daily on Day 1, 3 mg twice daily on Day 2, 6 mg twice daily on Day 3).
In Study 4, the primary endpoint was change in YMRS total score from baseline to Day 28.
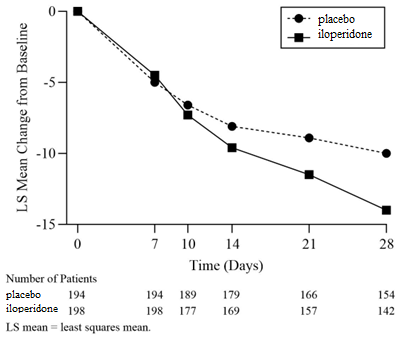
The iloperidone group was superior to the placebo group on the primary endpoint. Examination of subgroups did not reveal clear evidence of differential responsiveness on the basis of age, sex, or race. The results of Study 4 are shown in Table 8, and the LS mean changes from baseline in YMRS total score are shown in Figure 3.
Table 8. Primary Efficacy Results: Change from Baseline in YMRS Total Score to Day 28 in the Acute Treatment of Manic or Mixed Episodes Associated with Bipolar I Disorder in Adults (Study 4):
| Treatment Group (# ITT patients) | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Differencea (95% CI) |
| Iloperidone* (n=198) | 29.2 (5.27) | -14.0 (0.64) | -4.0 (-5.70, -2.25) |
| Placebo (n=194) | 28.8 (4.64) | -10.0 (0.63) |
ITT = intent-to-treat, YMRS = Young Mania Rating Scale, LS mean = least Squares mean, SD = standard deviation, SE = standard error
* In Study 4, patients who received iloperidone, received 12 mg twice daily (24 mg/day) after a four-day titration. If the iloperidone-treated patients were CYP2D6 poor metabolizers, they received 6 mg twice daily (12 mg/day)) after a two-day titration.
a Difference (iloperidone group minus placebo group) in least-squares mean change from baseline Iloperidone group was superior to the placebo group
Figure 3. Change from Baseline in YMRS Total Score by Study Visit in the Acute Treatment of Manic or Mixed Episodes Associated with Bipolar I Disorder in Adults (Study 4):
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.