EZETAST Film-coated tablet Ref.[49962] Active ingredients: Atorvastatin Ezetimibe
Source: Pharmaceutical Benefits Scheme (AU) Revision Year: 2021 Publisher: Lupin Australia Pty Ltd, Suite 2, Level 2, 19-23 Prospect Street, Box Hill, VIC, 3128 Distributor: Arrotex Pharmaceuticals Pty Ltd, 15-17 Chapel Street, Cremorne, VIC, 3121, Australia
5.1. Pharmacodynamic properties
Mechanism of Action
Ezetimibe and atorvastatin tablet is a lipid-lowering product that selectively inhibits the intestinal absorption of cholesterol and related plant sterols and inhibits the endogenous synthesis of cholesterol.
Plasma cholesterol is derived from intestinal absorption and endogenous synthesis. Ezetimibe and atorvastatin are lipid-lowering compounds with complementary mechanisms of action. Together these distinct mechanisms reduce total-C, LDL-C, Apo B, TG, and non-HDL-C, and increase HDL-C beyond either treatment alone, through dual inhibition of cholesterol absorption and synthesis.
Clinical studies demonstrate that elevated levels of total-C, LDL-C and Apo B, the major protein constituent of LDL, promote human atherosclerosis. In addition, decreased levels of HDL-C are associated with the development of atherosclerosis. Epidemiologic studies have established that cardiovascular morbidity and mortality vary directly with the level of total-C and LDL-C and inversely with the level of HDL-C. Like LDL, cholesterol-enriched triglyceride-rich lipoproteins, including very-lowdensity lipoproteins (VLDL), intermediate-density lipoproteins (IDL), and remnants, can also promote atherosclerosis.
Ezetimibe
Ezetimibe has a mechanism of action that differs from other classes of cholesterol reducing compounds (eg. statins, bile acid sequestrants [resins], fibric acid derivatives, and plant sterols.)
The molecular target of ezetimibe is the sterol transporter, Niemann-Pick C1-Like 1 (NPC1L1), which is responsible for the intestinal uptake of cholesterol and phytosterols. Ezetimibe therefore inhibits the absorption of cholesterol, leading to a decrease in the delivery of intestinal cholesterol to the liver. This causes a reduction of hepatic cholesterol stores and an increase in clearance of cholesterol from the blood. Ezetimibe does not increase bile acid excretion (like bile acid sequestrants) and does not inhibit cholesterol synthesis in the liver (like statins).
In a 2 week clinical study in 18 hypercholesterolaemic patients, ezetimibe inhibited intestinal cholesterol absorption by 54%, compared with placebo. A series of preclinical studies was performed to determine the selectivity of ezetimibe for inhibiting cholesterol absorption. Ezetimibe inhibited the absorption of 14C]-cholesterol with no effect on the absorption of triglycerides, fatty acids, bile acids, progesterone, ethinyloestradiol, or the fat soluble vitamins A and D.
Atorvastatin
Atorvastatin is a synthetic lipid-lowering agent. Atorvastatin is an inhibitor of HMG-CoA reductase, the rate-limiting enzyme that converts 3-hydroxy-3-methyl-glutaryl-coenzyme A to mevalonate, a precursor of sterols, including cholesterol. Triglycerides (TG) and cholesterol in the liver are incorporated into verylow-density lipoprotein (VLDL) and released into the plasma for delivery to peripheral tissues. Lowdensity lipoprotein (LDL) is formed from VLDL and is catabolised primarily through the high affinity LDL receptor.
Atorvastatin lowers plasma cholesterol and lipoprotein levels by inhibiting HMG-CoA reductase and cholesterol synthesis in the liver and by increasing the number of hepatic LDL receptors on the cellsurface to enhance uptake and catabolism of LDL. Atorvastatin reduces LDL production and the number of LDL particles. Atorvastatin produces a marked and sustained increase in LDL receptor activity coupled with a beneficial change in the quality of circulating LDL particles.
Atorvastatin reduces total-C, LDL-C, and Apo B in both normal volunteers and in patients with homozygous and heterozygous familial hypercholesterolaemia (FH), non-familial forms of hypercholesterolaemia, and mixed dyslipidaemia. Atorvastatin also reduces very low-density lipoprotein cholesterol (VLDL-C) and TG and produces variable increases in HDL-C and apolipoprotein A-1. Atorvastatin reduces total-C, LDL-C, VLDL-C, Apo B and TG, and increases HDL-C in patients with isolated hypertriglyceridaemia. Atorvastatin reduces intermediate-density lipoprotein cholesterol (IDL-C) in patients with dysbetalipoproteinaemia.
In animal models, atorvastatin limits the development of lipid-enriched atherosclerotic lesions and promotes the regression of pre-established atheroma.
Atorvastatin and its metabolites are responsible for pharmacological activity in humans. The liver is its primary site of action and the principal site of cholesterol synthesis and LDL clearance. Drug dose rather than systemic drug concentration correlates better with LDL-C reduction. Individualisation of drug dose should be based on therapeutic response (see Section 4.2 Dose and Method of Administration).
Clinical Trials
In controlled clinical studies, co-administration of ezetimibe and atorvastatin significantly reduced total cholesterol (total-C), low-density lipoprotein cholesterol (LDL-C), apolipoprotein B (Apo B), triglycerides (TG), and non-high-density lipoprotein cholesterol (non-HDL-C), and increased high-density lipoprotein cholesterol (HDL-C) in patients with hypercholesterolaemia.
Primary Hypercholesterolaemia
Ezetimibe Initiated Concurrently with Atorvastatin
In a multicentre, double-blind, placebo-controlled, clinical study (P0692) in patients with hyperlipidaemia, 628 patients (260 male, 368 female) were treated for up to 12 weeks and 246 for up to an additional 48 weeks. Patients were 18 to 86 years of age, with baseline LDL-C concentrations between 130 to 253 mg/dL (3.37 to 6.55 mmol/L) (mean baseline LDL-C ranged from 175 and 184 mg/dL [4.53 and 4.77 mmol/L] across treatment groups). Sixty-three percent had risk factors or a history of cardiovascular disease. Patients were randomised to receive placebo, ezetimibe (10 mg), atorvastatin (10 mg, 20 mg, 40 mg, or 80 mg), or co-administered ezetimibe and atorvastatin equivalent to ezetimibe/atorvastatin (10/10, 10/20, 10/40, and 10/80 mg) in the 12 week study. After completing the 12 week study, eligible patients were assigned to co-administered ezetimibe and atorvastatin equivalent to Ezetimibe/Atorvastatin (10/10-10/80 mg) or atorvastatin (10-80 mg/day) for an additional 48 weeks (see Section 5.1 Pharmacodynamic Properties – Clinical Trials, Long-term studies, P2154).
Eight percent of subjects discontinued treatment early, 5% were due to adverse events. There was no trend across treatment groups in the distribution of subjects who discontinued or in the reasons for discontinuation.
Patients receiving all doses of ezetimibe and atorvastatin tablets were compared to those receiving all doses of atorvastatin. The primary endpoint was percent change from baseline in direct LDL-C at study endpoint (12 weeks). Secondary endpoints were percent change from baseline in calculated LDL-C, TC, TG, HDL-C and Apo B at endpoint. Ezetimibe and atorvastatin tablets lowered total-C, LDL-C, Apo B, TG, and non-HDL-C, and increased HDL-C significantly more than atorvastatin alone (see Table 4).
Table 4. Response to Ezetimibe/Atorvastatin in Patients with Primary Hyperlipidaemia (ITT analysis) (Meana % change from Untreated Baselineb at 12 weeks):
| Treatment (Daily Dose) | N | Total-C | LDL-C | Apo B | TGa | HDL-C | Non-HDL-C |
|---|---|---|---|---|---|---|---|
| Pooled data (All Ezetimibe/Atorvastatin doses)c | 255 | -41 | -56 | -45 | -33 | +7 | -52 |
| Pooled data (All atorvastatin doses)c | 248 | -32 | -44 | -36 | -24 | +4 | -41 |
| Ezetimibe 10 mg | 65 | -14 | -20 | -15 | -5 | +4 | -18 |
| Placebo | 60 | +4 | +4 | +3 | -6 | +4 | +4 |
| Ezetimibe/Atorvastatin by dose | |||||||
| 10/10 | 65 | -38 | -53 | -43 | -31 | +9 | -49 |
| 10/20 | 62 | -39 | -54 | -44 | -30 | +9 | -50 |
| 10/40 | 65 | -42 | -56 | -45 | -34 | +5 | -52 |
| 10/80 | 63 | -46 | -61 | -50 | -40 | +7 | -58 |
| Atorvastatin by dose | |||||||
| 10 mg | 60 | -26 | -37 | -28 | -21 | +6 | -34 |
| 20 mg | 60 | -30 | -42 | -34 | -23 | +4 | -39 |
| 40 mg | 66 | -32 | -45 | -37 | -24 | +4 | -41 |
| 80 mg | 62 | -40 | -54 | -46 | -31 | +3 | -51 |
a For triglycerides, median % change from baseline.
b Baseline – on no lipid-lowering drug.
c Ezetimibe and Atorvastatin tablets pooled (10/10-10/80) significantly reduced total-C, LDL-C, Apo B, TG, non-HDLC, and significantly increased HDL-C compared to all doses of atorvastatin pooled (10-80 mg).
The changes in lipid endpoints after an additional 48 weeks of treatment with ezetimibe and atorvastatin tablets (all doses) or with atorvastatin (all doses) were generally consistent with the 12 week data displayed above.
Ezetimibe Added to Stable Atorvastatin Therapy
In a multicentre, double-blind, placebo-controlled, 8 week study (P2173/2246), 769 (443 male, 326 female) patients aged 22 to 85 years with hypercholesterolaemia (baseline LDL-C ranged from 71 to 455 mg/dL [1.84 to 11.78 mmol/L]; mean baseline LDL-C 138 to 139 mg/dL [3.57 to 3.60 mmol/L] across the treatment groups), already receiving statin monotherapy and not at National Cholesterol Education Program (NCEP) LDL-C goal (2.59 to 4.14 mmol/L, depending on baseline characteristics) were randomised to receive either ezetimibe 10 mg or placebo in addition to their on-going statin therapy. Sixty-eight percent of subjects had CHD, diabetes and/or CHD equivalent disease with LDL-C ≥100 mg/dL (≥2.59 mmol/L).
Fifty-three subjects discontinued study treatment early, 34 were due to adverse events. There was no trend across treatment groups in the distribution of subjects who discontinued or the reasons for discontinuation.
The primary efficacy endpoint was the difference in mean percent change in LDL-C between the treatment groups. The secondary endpoints included the percentage of subjects who achieved NCEP ATP II target LDL-C levels. Endpoints were analysed for a modified ITT population (all subjects who received randomised treatment and had at least one post-baseline value).
Three percent of subjects discontinued treatment early due to adverse events in each treatment group.
In the subgroup of 308 patients with hypercholesterolaemia already receiving atorvastatin monotherapy and not at LDL-C goal at baseline (~83%), significantly more patients randomised to ezetimibe co-administered with atorvastatin achieved their LDL-C goal at study endpoint compared to patients randomised to placebo co-administered with atorvastatin, 72% vs. 27%; the analysis was post-hoc. Ezetimibe added to atorvastatin therapy lowered LDL-C significantly more than placebo added to atorvastatin therapy, 25% vs. 4%. In addition, ezetimibe added to atorvastatin therapy significantly decreased total-C, Apo B, and TG compared with placebo added to atorvastatin therapy.
After 8 weeks of treatment, 730 patients had their blinded ezetimibe or placebo withdrawn and were continued on their stable statin therapy for another 6 weeks (P2173R). Twenty-one subjects discontinued treatment during the reversibility phase. Lipid parameters were observed to return to their pre-treatment values during this period, without any evidence of rebound.
Another double-blind, randomised, placebo-controlled study (P040) evaluated the effect of ezetimibe 10 mg/day added to ongoing statin therapy vs. continued statin therapy alone (at unchanged dose) in 3,030 patients (52% male) mean age 62 years and with hypercholesterolemia who were not at their NCEP ATP III Target LDL-C level. Mean baseline LDL-C was 129 mg/dL (3.34 mmol/L). Approximately 78% of patients had CHD or risk equivalent. Adverse experiences resulting in discontinuation occurred in 2.1% of the statin monotherapy groups and in 1.4% of the ezetimibe/statin groups.
The primary outcome was percent change in LDL-C from baseline at week 6. In the subgroup of patients receiving atorvastatin (n=1194) the addition of ezetimibe to atorvastatin produced a reduction of 27.2% in LDL-C at week 6 (relative to the on-statin baseline) compared to 4.2% for placebo, a difference of 23.0% (Modified ITT analysis – excluded patients who had adverse clinical or laboratory experiences, lost to follow-up, protocol deviations, withdrawn consent, discontinued for other reasons and missing LDL-C measurements). In addition, a greater number of patients in the active ezetimibe group achieved their NCEP ATP III Target Goal for LDL-C, 23.9% for atorvastatin alone vs. 74.6% for ezetimibe + atorvastatin (secondary outcome).
Ezetimibe Add-on to On-going Atorvastatin Therapy (Titration Studies)
A multicentre, double-blind, controlled, 14 week study (P00693) was conducted in 621 patients (330 male, 291 female) with heterozygous familial hypercholesterolemia (HeFH), coronary heart disease (CHD), or multiple cardiovascular risk factors (≥2), adhering to an National Cholesterol Education Program (NCEP) Step I or stricter diet. Patients were 18 to 82 years of age with baseline LDL-C of 117 to 466 mg/dL (3.03 to 12.07 mmol/L) (mean LDL-C: 186 mg/dL and 187 mg/dL [4.82 mmol/L and 4.84 mmol/L] for patients receiving co-administered ezetimibe and atorvastatin 10/10 and atorvastatin 20 mg respectively). Fifty-eight percent of patients were diagnosed with HeFH and the majority of subjects (87%) had risk factors or a family history of cardiovascular disease.
All patients received atorvastatin 10 mg for a minimum of 4 weeks prior to randomisation. Patients were then randomised to receive either co-administered ezetimibe and atorvastatin (equivalent to ezetimibe/atorvastatin 10/10) or atorvastatin 20 mg/day monotherapy. Patients who did not achieve their LDL-C target goal after 4 and/or 9 weeks of randomised treatment were titrated to double the atorvastatin dose. There were 181 patients in the atorvastatin monotherapy treatment arm (all doses) and 181 in the co-administration arm (all doses).
Nine percent of subjects discontinued treatment early, 4% due to adverse events. There was no trend across treatment groups in the distribution of subjects who discontinued or in the reasons for discontinuation.
Efficacy analyses were carried out on an ITT basis.
The primary endpoint was proportion of subjects achieving target LDL-C levels of ≤2.59 mmol/L (≤100 mg/dL) at week 14. A higher proportion of subjects on ezetimibe and atorvastatin tablets (22%), than on atorvastatin alone (7%) achieved target LDL-C levels of ≤2.59 mmol/L (100 mg/dL) at week 14 (p<0.01).
The secondary endpoints included mean percent change from baseline in LDL-C and proportion of subjects achieving target LDL-C levels at week 4. Ezetimibe/atorvastatin 10/10 was significantly more effective than doubling the dose of atorvastatin to 20 mg in further reducing total-C, LDL-C, TG, and nonHDL-C. Results for HDL-C between the two treatment groups were not significantly different (see Table 5.) In addition, at week 4 significantly more patients receiving ezetimibe/atorvastatin 10/10 attained LDL-C <2.6 mmol/L (<100 mg/dL) compared to those receiving atorvastatin 20 mg, 12% vs. 2%. The baseline mean LDL-C levels for patients receiving ezetimibe/atorvastatin 10/10 and atorvastatin 20 mg were 186 mg/dL and 187 mg/dL, respectively.
Table 5. Response to Ezetimibe and Atorvastatin tablets after 4 Weeks in Patients with CHD or Multiple Cardiovascular Risk Factors and an LDL-C ≥130 mg/dL (≥3.37 mmol/L) (Modified ITT analysis) (Mean* % Change from Baseline†):
| Treatment (Daily Dose) | N | Total-C | LDL-C | HDL-C | TG* | Non-HDL-C |
|---|---|---|---|---|---|---|
| Ezetimibe/atorvastatin 10/10 | 305 | -17 | -24‡ | +2 | -9‡ | -22‡ |
| Atorvastatin 20 mg | 316 | -6 | -9 | +1 | -4 | -8 |
* For triglycerides, median % change from baseline.
† Patients on atorvastatin 10 mg, then switched to ezetimibe and atorvastatin tablets 10/10 or titrated to atorvastatin 20 mg.
‡ p<0.05 for difference with atorvastatin.
The Titration of Atorvastatin Versus Ezetimibe Add-On to Atorvastatin in Patients with Hypercholesterolaemia (TEMPO) study, a multicentre, double-blind, controlled, 6 week study (P079), included 184 patients (55% male) mean age 57 (range 24 to 78 years) with an LDL-C level ≥2.6 mmol/L and ≤4.1 mmol/L (≥100 mg/dL and ≤160 mg/dL), mean baseline LDL-C of 3.08 mmol/L (118.9 mg/dL) and at moderate high risk for coronary heart disease (CHD). All patients received atorvastatin 20 mg for a minimum of 4 weeks prior to randomisation. Patients not at the optional NCEP ATP III LDL-C level (<2.6 mmol/L [<100 mg/dL]) were randomised to receive either co-administered ezetimibe and atorvastatin (equivalent to ezetimibe/atorvastatin 10/20) or atorvastatin 40 mg for 6 weeks. Thirteen patients discontinued study treatment; 2 were due to adverse events.
The primary endpoint was percent change from baseline LDL-C at week 6. Efficacy was evaluated for all patients who had at least one dose of study medication, had a baseline measurement and at least one post-baseline measurement (modified ITT). Ezetimibe/atorvastatin 10/20 was significantly more effective than doubling the dose of atorvastatin to 40 mg in further reducing total-C, LDL-C, Apo B and non-HDL-C. Results for HDL-C and TG between the two treatment groups were not significantly different (see Table 6). In addition, significantly more patients receiving ezetimibe/atorvastatin 10/20 attained LDL-C <2.6 mmol/L (<100 mg/dL) compared to those receiving atorvastatin 40 mg, 84% vs. 49% (secondary endpoint).
Table 6. Response to Ezetimibe/Atorvastatin in Patients with Primary Hypercholesterolaemia (Modified ITT analysis) (Meana % Change from Baselineb):
| Treatment (Daily Dose) | N | Total-C | LDL-C | Apo B | HDL-C | TGa | Non-HDL-C |
|---|---|---|---|---|---|---|---|
| Ezetimibe/atorvastatin 10/20 | 92 | -20c | -31c | -21c | +3 | -18 | -27c |
| Atorvastatin 40 mg | 92 | -7 | -11 | -8 | +1 | -6 | -10 |
a For triglycerides, median % change from baseline.
b Patients on atorvastatin 20 mg, then switched to ezetimibe/atorvastatin 10/20 or titrated to atorvastatin 40 mg.
c p<0.05 for difference with atorvastatin.
The Ezetimibe Plus Atorvastatin Versus Atorvastatin Titration in Achieving Lower LDL-C Targets in Hypercholesterolemic Patients (EZ-PATH) study, a multicentre, double-blind, controlled, 6 week study (P090), included 556 (60% male) patients with a mean age of 61 years and an LDL-C level ≥1.8 mmol/L and ≤4.1 mmol/L (≥70 mg/dL and ≤160 mg/dL) and at high risk for coronary heart disease (CHD). All patients received atorvastatin 40 mg for a minimum of 4 weeks prior to randomisation. Patients not at the optional NCEP ATP III LDL-C level <1.8 mmol/L (<70 mg/dL) were randomised to receive either co-administered ezetimibe and atorvastatin (equivalent to ezetimibe/atorvastatin 10/40) or atorvastatin 80 mg for 6 weeks. Four patients in the ezetimibe/atorvastatin group and 6 patients in the atorvastatin monotherapy group experienced an adverse event that lead to discontinuation of the study treatment.
The primary outcome was mean percent change from baseline in LDL-C at week 6. Efficacy was evaluated for all patients who had at least one dose of study medication, had a baseline measurement and at least one post-baseline measurement (modified ITT). Ezetimibe/atorvastatin 10/40 was significantly more effective than doubling the dose of atorvastatin to 80 mg in further reducing total-C, LDL-C, Apo B, TG, and non-HDL-C. Results for HDL-C between the two treatment groups were not significantly different (see Table 7). In addition, significantly more patients receiving ezetimibe/atorvastatin 10/40 attained LDL-C <1.8 mmol/L (<70 mg/dL) compared to those receiving atorvastatin 80 mg, 74% vs. 32% (secondary endpoint).
Table 7. Response to Ezetimibe and Atorvastatin tablets in Patients with Primary Hypercholesterolaemia (Modified ITT analysis) (Meana % Change from Baselineb):
| Treatment (Daily Dose) | N | Total-C | LDL-C | Apo B | HDL-C | TGa | Non-HDL-C |
|---|---|---|---|---|---|---|---|
| Ezetimibe/atorvastatin 10/40 | 277 | -17c | -27c | -18c | 0 | -12c | -23c |
| Atorvastatin 80 mg | 279 | -7 | -11 | -8 | -1 | -6 | -9 |
a For triglycerides, median % change from baseline.
b Patients on atorvastatin 40 mg, then switched to ezetimibe/atorvastatin 10/40 or titrated to atorvastatin 80 mg.
c p<0.05 for difference with atorvastatin.
A multicentre, randomised, double-blind, parallel-arm, 12 week study (P112) evaluated the lipid altering efficacy and safety of the addition of ezetimibe 10 mg to atorvastatin 10 mg, as compared to doubling the dose of atorvastatin from 10 mg to 20 mg and followed by further up-titration from atorvastatin 20 to 40 mg. The 1053 patients (53.3% female) were 65 years of age and older (mean age 71.2; range 65 to >90 years), at high risk for CHD with or without diagnosed atherosclerotic vascular disease (AVD) who had not reached an LDL-C level of <70 mg/dL (1.81 mmol/L) or <100 mg/dL (2.59 mmol/L), respectively, and on atorvastatin 10 mg/day. Mean baseline LDL-C levels were 102 mg/dL (2.64 mmol/L). Twenty-two patients (2%) discontinued treatment due to an adverse event.
The primary endpoint was percent change in LDL-C from baseline to week 6. Efficacy was evaluated for all patients who had at least one dose of study medication, had a baseline measurement and at least one post-baseline measurement (modified ITT). Ezetimibe added to atorvastatin (equivalent to ezetimibe/atorvastatin 10/10) significantly reduced LDL-C from baseline after 6 weeks of treatment compared with doubling the dose of atorvastatin from 10 to 20 mg (26.7% vs. -12.8%; p<0.001). Additionally, treatment with ezetimibe/atorvastatin 10/10 resulted in a significantly greater percentage of patients achieving LDL-C <100 mg/dL (2.59 mmol/L) for high risk patients without AVD and <70 mg/dL (1.81 mmol/L) for high risk patients with AVD after 12 weeks of treatment, compared to the group that had atorvastatin increased to 40 mg at week 6 (49.4% vs. 39.3%, p<0.001).
Switching Study
In a multicentre, double-blind, controlled, 12 week, 2-phase study (P162), 1,539 high-cardiovascular-risk patients, with a LDL-C level between 2.6 mmol/L and 4.1 mmol/L (100 and 160 mg/dL) at baseline, on atorvastatin 10 mg daily were randomised to one of three treatment groups: two of which were atorvastatin 20 mg or ezetimibe/atorvastatin 10/10. After 6 weeks of treatment (Phase I), based on a random allocation schedule established at the start of Phase I, patients taking atorvastatin 20 mg who failed to achieve a LDL-C level <2.6 mmol/L (100 mg/dL) were switched to either atorvastatin 40 mg or ezetimibe/atorvastatin 10/20 for 6 weeks (Phase II). Reductions in LDL-C and comparisons between the ezetimibe/atorvastatin group and other treatment groups studied are shown in Table 8.
Table 8. Response to Ezetimibe and Atorvastatin tablets (Co-administration of Ezetimibe and atorvastatin) in High-Risk Patients with a LDL-C Level Between 2.6 mmol/L and 4.1 mmol/L (100 and 160 mg/dL) on Atorvastatin 10 mg Daily at Baseline:
| Treatment | N | Percent Change from Baseline† | |||||
|---|---|---|---|---|---|---|---|
| Total-C | LDL-C | Apo B | TG‡ | HDL-C | Non-HDL-C | ||
| Phase I Switched from atorvastatin 10 mg | |||||||
| Ezetimibe/atorvastatin 10/10 | 120 | -13.5 | -22.2 | -11.3 | -6.0 | +0.6 | -18.3 |
| Atorvastatin 20 mg | 480 | -6.4§ | -9.5§ | -6.0¶ | -3.9 | -1.1 | -8.1§ |
| Phase II Switched from atorvastatin 20 mg | |||||||
| Ezetimibe/atorvastatin 10/20 | 124 | -10.7 | -17.4 | -9.8 | -5.9 | +0.7 | -15.1 |
| Atorvastatin 40 mg | 124 | -3.8Þ | -6.9Þ | -5.4 | -3.1 | +1.7 | -5.8Þ |
† M-Estimates(based on the method of Huber; 95% CI and p-value were obtained from fitting a robust Regression model with terms for treatment and baseline).
‡ Geometric mean percent changes from baseline in TG were calculated based on back-transformation via exponentiation of the model-based least square (LS) means and expressed as (geometric mean – 1) multiplied by 100.
^§ p<0.001 versus Ezetimibe/Atorvastatin 10/10.
¶ p<0.01 versus Ezetimibe/Atorvastatin 10/10.
Þ p<0.001 versus Ezetimibe/Atorvastatin 10/20.
Table 8 does not contain data comparing the effects of ezetimibe/atorvastatin 10/10 or 10/20 to doses higher than atorvastatin 40 mg.
Long-term studies
A 12 month, blinded comparator study (P2154) enrolled 246 (101 male, 145 female) subjects who had completed study P0692. Patients in this follow-on study were aged from 26 to 86 years, with primary hypercholesterolaemia. Mean baseline LDL-C was 184.6 and 180.6 mg/dL (4.78 and 4.68 mmol/L) in the atorvastatin monotherapy and co-administration groups respectively. A greater proportion of subjects in the monotherapy group had a medical history or physical finding of cardiovascular disease (31% vs. 19%) and were hypertensive (42% vs. 34%) compared to the co-administration group. Forty-one subjects discontinued treatment; 22 discontinued due to adverse events (3/45 in the monotherapy group and 19/201 in the co-administration group).
Patients were initially dosed with either double-blind ezetimibe 10 mg or matching placebo co-administered with open-label atorvastatin 10 mg once daily in the morning. After at least 6 weeks, the atorvastatin dose could be titrated up incrementally to a maximum of 80 mg once daily to achieve the subject’s NCEP ATP II target LDL-C level. Efficacy evaluations were performed on all subjects in the follow-up study who had at least one post-baseline lipid measurement (modified ITT). Overall, co-administration of ezetimibe and atorvastatin reduced LDL-C levels during this 12 month study significantly more than atorvastatin monotherapy. At week 6 (the first time point assessed), LDL-C was reduced from baseline by approximately 37% in the atorvastatin monotherapy group and by approximately 53% in the ezetimibe/atorvastatin group. The LDL-C-lowering effect was seen by six weeks of treatment and maintained during the 12 month double-blind study period.
A 12 month, open-label study (P1418) was conducted in patients with HeFH, known CHD or multiple cardiovascular risk factors (≥ 2) who were not controlled by a starting dose of atorvastatin 10 mg and had successfully completed a 14 week double-blind efficacy and safety study (P00693). Four hundred and thirty-two hypercholesterolaemic patients (56% male) with mean age 52 years (range 18 to 82 years) and mean baseline LDL-C 187 mg/dL (4.84 mmol/L) received open-label ezetimibe 10 mg co-administered with atorvastatin 10 mg (equivalent to ezetimibe/atorvastatin 10/10) at the beginning of the study, with up titration of atorvastatin to reach target LDL-C. Eighty-nine percent of patients had a cardiovascular risk factor or family history of cardiovascular disease. Approximately 38% had a history of hypertension and 26% had angina pectoris. Thirty-four subjects discontinued from the follow-on study, 12 due to adverse events.
Efficacy evaluations were carried out on all subjects in the follow-on study who had at least one postbaseline lipid measurement (modified ITT). Over the 12 month study period, ezetimibe/atorvastatin 10/10-10/80 was effective in achieving and maintaining a reduction in LDL-C. The mean LDL-C value at study end was reduced by 30% from the parent study baseline. Reductions were noted as of month 1, were slightly greater at month 3 and were maintained at similar levels throughout the study period. Commensurate reductions in TC were observed, and reductions in TG, and an increase in HDL-C, were also noted over time.
Homozygous Familial Hypercholesterolaemia (HoFH)
A double-blind, randomised, 12 week study (P1030) was performed in 50 patients (21 male, 29 female, aged 11 to 74 years of age) with a clinical and/or genotypic diagnosis of HoFH. Baseline LDL-C concentrations ranged from 116 to 652 mg/dL (3.00 to 16.89 mmol/L) (mean: 346 mg/dL [8.96 mmol/L] in the monotherapy group; 321 mg/dL [8.31 mmol/L] in the co-administration group). Approximately 74% of subjects had known family history of coronary artery disease and approximately 16% had some degree of hypertension at baseline. Twenty-five subjects received concomitant apheresis or plasmapheresis. Two subjects discontinued treatment early due to adverse events considered to be unrelated to study treatment.
Data were analysed from a subgroup of patients (n=36) receiving atorvastatin 40 mg at baseline (ITT). The primary endpoint was the percent change from baseline in direct LDL-C concentration at week 12. Increasing the dose of atorvastatin from 40 to 80 mg (n=12) produced a reduction of LDL-C of 2% from baseline on atorvastatin 40 mg. Co-administered ezetimibe and atorvastatin equivalent to ezetimibe/atorvastatin (10/40 and 10/80 pooled, n=24), produced a reduction of LDL-C of 19% from baseline on atorvastatin 40 mg. In those patients co-administered ezetimibe and atorvastatin equivalent to ezetimibe/atorvastatin (10/80 mg, n=12), a reduction of LDL-C of 25% from baseline on atorvastatin 40 mg was produced.
After completing the 12 week study, eligible patients (n=35), who were receiving atorvastatin 40 mg at baseline, were assigned to co-administered ezetimibe and atorvastatin equivalent to ezetimibe/atorvastatin 10/40 for up to an additional 24 months (P1417). Following at least 4 weeks of treatment, the atorvastatin dose could be doubled to a maximum dose of 80 mg. One patient discontinued treatment due to a drug related adverse event. At the end of the 24 months, ezetimibe/atorvastatin (10/40 and 10/80 mg pooled) produced a reduction of 18% in LDL-C that was consistent with that seen in the 12 week study (modified ITT analysis – included patients who completed 24 months of treatment).
Ezetimibe
In two multicentre, double-blind, placebo-controlled, 12 week studies in 1,719 patients with primary hypercholesterolaemia, ezetimibe significantly lowered total-C (-13%), LDL-C (-19%), Apo B (-14%), and TG (-8%), and increased HDL-C (+3%) compared to placebo. Reduction in LDL-C was consistent across age, sex, race and baseline LDL-C. In addition, ezetimibe had no effect on the plasma concentrations of the fat soluble vitamins A, D, and E, had no effect on prothrombin time, and did not impair adrenocortical steroid hormone production.
Atorvastatin
In a placebo-controlled study, the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT), the effect of atorvastatin 10 mg on fatal and non-fatal coronary heart disease was assessed in 10,305 hypertensive patients, 40-80 years old, with TC levels ≤ 251 mg/dL (6.5 mmol/L) and at least three cardiovascular risk factors. Patients were followed for a median duration of 3.3 years. Atorvastatin 10 mg significantly (p=0.0005) reduced the rate of coronary events (either fatal coronary heart disease [46 events in the placebo group vs. 40 events in the atorvastatin group] or nonfatal MI [108 events in the placebo group vs. 60 events in the atorvastatin group]) by 36% (based on incidences of 1.9% for atorvastatin vs. 3.0% for placebo).
Although this difference was statistically significant for the whole trial population, this difference was not statistically significant in specified subgroups such as diabetes, patients with left ventricular hypertrophy (LVH), previous vascular disease or metabolic syndrome.
There was no statistically significant reduction in the rate of total mortality, cardiovascular mortality or heart failure in the atorvastatin treated group compared to placebo.
Experience in non-Caucasians is limited and does not permit a precise estimate of the magnitude of the effects of ezetimibe and atorvastatin tablets.
Prevention of Cardiovascular Disease
To date, no clinical trials have been undertaken that demonstrate an improvement in cardiovascular outcome when a combination of ezetimibe and atorvastatin is used compared to atorvastatin alone. Indirect evidence for prevention of cardiovascular disease has been derived from a trial of ezetimibe in combination with simvastatin (the IMPROVE-IT trial) (details below). The magnitude of the incremental benefit achieved with the use of ezetimibe in addition to atorvastatin is assumed to be similar to that achieved using simvastatin and ezetimibe in the IMPROVE-IT trial. The effect of atorvastatin on the risk of cardiovascular events has been demonstrated in multiple cardiovascular outcomes studies including the ASCOT study (see Section 5.1 Pharmacodynamic Properties-Clinical Trials, Atorvastatin).
Ezetimibe in combination with simvastatin has been shown in the IMPROVE-IT trial to reduce the major cardiovascular events of non-fatal myocardial infarction and stroke in patients with coronary heart disease and a history of Acute Coronary Syndrome. Total mortality, cardiovascular mortality and rates of unstable angina requiring hospitalization and all coronary revascularization were unchanged. There was a small increase in the rate of haemorrhagic stroke that was not statistically significant.
The IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) was a multicentre, randomised, double-blind, active-control study of 18,144 patients enrolled within 10 days of hospitalisation for acute coronary syndrome (ACS; either acute myocardial infarction [MI] or unstable angina [UA]). Patients had an LDL-C ≤3.2 mmol/L (≤125 mg/dL) at the time of presentation with ACS if they had not been taking lipid-lowering therapy, or ≤2.6 mmol/L (≤100 mg/dL) if they had been receiving lipid-lowering therapy. All patients were randomised in a 1:1 ratio to receive either ezetimibe/simvastatin 10/40 mg (n=9067) or simvastatin 40 mg (n=9077) and followed for a median of 6.0 years.
Patients had a mean age of 63.6 years; 76% were male, 84% were Caucasian, and 27% were diabetic. The average LDL-C value at the time of study qualifying event was 2.1 mmol/L (80 mg/dL) for those on lipid-lowering therapy (n=6390) and 2.6 mmol/L (101 mg/dL) for those not on previous lipid-lowering therapy (n=11594). Prior to the hospitalization for the qualifying ACS event, 34% of the patients were on statin therapy. At one year, the average LDL-C for patients continuing on therapy was 1.4 mmol/L (53.2 mg/dL) for the ezetimibe/simvastatin group and 1.8 mmol/L (69.9 mg/dL) for the simvastatin monotherapy group. Lipid values were generally obtained for patients who remained on study therapy.
The primary endpoint was a composite consisting of cardiovascular death, major coronary events (MCE; defined as non-fatal myocardial infarction, documented unstable angina that required hospitalisation, or any coronary revascularisation procedure occurring at least 30 days after randomised treatment assignment) and non-fatal stroke. The study demonstrated that treatment with ezetimibe/simvastatin provided incremental benefit in reducing the primary composite endpoint of cardiovascular death, MCE, and non-fatal stroke compared with simvastatin alone (relative risk reduction of 6.4%, p=0.016). The primary endpoint occurred in 2,572 of 9,067 patients (7 year Kaplan-Meier [KM] rate 32.72%) in the ezetimibe/simvastatin group and 2,742 of 9,077 patients (7 year KM rate 34.67%) in the simvastatin alone group. (see Figure 1 and Table 9.) This incremental benefit is expected to be similar with co-administration of ezetimibe and atorvastatin. Total mortality was unchanged in this high-risk group (see Table 9).
There was an overall benefit for all strokes; however, there was a small non-significant increase in haemorrhagic stroke in the ezetimibe/simvastatin group compared with simvastatin alone (see Table 9). The risk of haemorrhagic stroke for ezetimibe co-administered with higher potency statins in long-term outcome studies has not been evaluated.
The treatment effect of ezetimibe/simvastatin was generally consistent with the overall results across many subgroups, including sex, age, race, medical history of diabetes mellitus, baseline lipid levels, prior statin therapy, prior stroke, and hypertension.
Figure 1. Effect of ezetimibe/simvastatin on the Primary Composite Endpoint of Cardiovascular Death, Major Coronary Event, or Non-fatal Stroke:
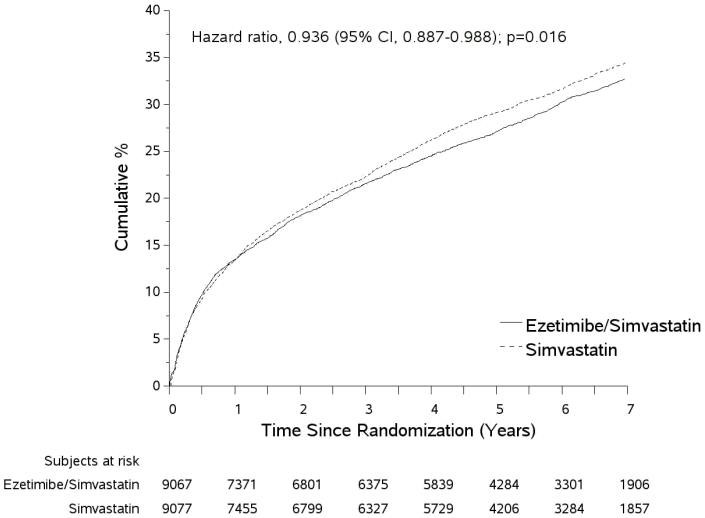
Table 9. Major Cardiovascular Events by Treatment Group in All Randomised Patients in IMPROVE-IT:
| Outcome | Ezetimibe/Simvastatin 10/40 mg* (N=9067) | Simvastatin 40 mg† (N=9077) | Hazard Ratio (95% CI) | p-value | ||
|---|---|---|---|---|---|---|
| n | K-M %‡ | n | K-M %‡ | |||
| Primary Composite Efficacy Endpoint | ||||||
| CV death, Major Coronary Events and non-fatal stroke) | 2572 | 32.72% | 2742 | 34.67% | 0.936 (0.887, 0.988) | 0.016 |
| Components of Primary Composite Endpoint and Select Efficacy Endpoints (first occurrences of specified event at any time) | ||||||
| Cardiovascular death | 537 | 6.89% | 538 | 6.84% | 1.000 (0.887, 1.127) | 0.997 |
| Major Coronary Event | ||||||
| Non-fatal MI | 945 | 12.77% | 1083 | 14.41% | 0.871 (0.798, 0.950) | 0.002 |
| Unstable angina requiring hospitalisation | 156 | 2.06% | 148 | 1.92% | 1.059 (0.846, 1.326) | 0.618 |
| Coronary revascularisation after 30 days | 1690 | 21.84% | 1793 | 23.36% | 0.947 (0.886, 1.012) | 0.107 |
| Non-fatal stroke | 245 | 3.49% | 305 | 4.24% | 0.802 (0.678, 0.949) | 0.010 |
* 6% were up-titrated to ezetimibe/simvastatin 10/80 mg.
† 27% were up-titrated to simvastatin 80 mg.
‡ Kaplan-Meier estimate at 7 years.
Other Studies
The use of ezetimibe with fenofibrate in patients with mixed hyperlipidaemia demonstrated a numerically higher incidence of cholecystectomies in patients in the co-administration group compared with those in the monotherapy groups (see Section 4.3 Contraindications and Section 4.8 Adverse Effects (Undesirable Effects)). Each drug contributed to lowering LDL-C, but the effects on triglycerides and HDL-C were related to fenofibrate and were not enhanced by co- administration. Longer term clinical outcomes such as mortality and morbidity were not investigated.
5.2. Pharmacokinetic properties
Ezetimibe/atorvastatin has been shown to be bioequivalent at the high and low end of dosage to co-administration of corresponding doses of ezetimibe and atorvastatin tablets. Bioequivalence at the mid dose ranges has been extrapolated.
The effects of a high-fat meal on the pharmacokinetics of ezetimibe and atorvastatin when administered as ezetimibe/atorvastatin tablets are comparable to those reported for the individual tablets.
Ezetimibe
Absorption
After oral administration, ezetimibe is rapidly absorbed and extensively conjugated to a pharmacologically active phenolic glucuronide (ezetimibe-glucuronide). Mean maximum plasma concentrations (Cmax) occur within 1 to 2 hours for ezetimibe-glucuronide and 4 to 12 hours for ezetimibe. The absolute bioavailability of ezetimibe cannot be determined as the compound is virtually insoluble in aqueous media suitable for injection.
Concomitant food administration (high-fat or non-fat meals) had no effect on the oral bioavailability of ezetimibe when administered as ezetimibe 10 mg tablets.
Distribution
Ezetimibe and ezetimibe-glucuronide are bound 99.7% and 88 to 92% to human plasma proteins, respectively.
Metabolism
Ezetimibe is metabolised primarily in the small intestine and liver via glucuronide conjugation (a phase II reaction) with subsequent biliary excretion. Minimal oxidative metabolism (a phase I reaction) has been observed in all species evaluated. Ezetimibe and ezetimibe-glucuronide are the major drug-derived compounds detected in plasma, constituting approximately 10 to 20% and 80 to 90% of the total drug in plasma, respectively. Both ezetimibe and ezetimibe-glucuronide are slowly eliminated from plasma with evidence of significant enterohepatic recycling. The half-life for ezetimibe and ezetimibeglucuronide is approximately 22 hours.
Excretion
Following oral administration of 14C-ezetimibe (20 mg) to human subjects, total ezetimibe accounted for approximately 93% of the total radioactivity in plasma. Approximately 78% and 11% of the administered radioactivity were recovered in the faeces and urine, respectively, over a 10-day collection period. After 48 hours, there were no detectable levels of radioactivity in the plasma.
Atorvastatin
Absorption
Atorvastatin is rapidly absorbed after oral administration; maximum plasma concentrations occur within 1 to 2 hours. A constant proportion of atorvastatin is absorbed intact. The absolute bioavailability is 14%. The low systemic availability is attributed to pre-systemic clearance in gastrointestinal mucosa and/or hepatic first-pass metabolism. Although food decreases the rate and extent of drug absorption by approximately 25% and 9%, respectively, as assessed by Cmax and AUC, LDL-C reduction is similar whether atorvastatin is given with or without food. Plasma atorvastatin concentrations are lower (approximately 30% for Cmax and AUC) following evening drug administration compared with morning. However, LDL-C reduction is the same regardless of the time of day of drug administration (see Section 4.2 Dose and Method of Administration).
Distribution
The mean volume of distribution of atorvastatin is about 400 litres. Atorvastatin is ≥98% bound to plasma proteins. A RBC/plasma ratio of approximately 0.25 indicates poor drug penetration into red blood cells. Based on observations in rats, atorvastatin is likely to be secreted in human milk (see Section 4.4 Special Warnings and Precautions for Use).
Metabolism
In humans, atorvastatin is extensively metabolised to ortho- and para-hydroxylated derivatives. In vitro inhibition of HMG-CoA reductase by ortho- and para-hydroxylated metabolites is equivalent to that of atorvastatin. Approximately 70% of circulating inhibitory activity for HMG-CoA reductase is attributed to active metabolites. In vitro studies suggest the importance of atorvastatin metabolism by cytochrome P450 3A4, consistent with increased plasma concentrations of atorvastatin in humans following co-administration with erythromycin, a known inhibitor of this isozyme (see Section 4.4 Special Warnings and Precautions for Use). In animals, the ortho-hydroxy metabolite undergoes further glucuronidation.
Excretion
Atorvastatin is eliminated primarily in bile following hepatic and/or extrahepatic metabolism. However, the drug does not appear to undergo enterohepatic recirculation. Mean plasma elimination half-life of atorvastatin in humans is approximately 14 hours. The half-life of inhibitory activity for HMG-CoA reductase is 20 to 30 hours due to the contribution of active metabolites. Less than 2% of a dose of atorvastatin is recovered in urine following oral administration.
Characteristics in Patients (Special Populations)
Paediatric patients
Ezetimibe
The absorption and metabolism of ezetimibe are similar between children and adolescents (≥10 years) and adults. Limited Pk data are available in children aged ≥6 to 10 years of age. Pharmacokinetic data in the paediatric population <6 years of age are not available.
Atorvastatin
Pharmacokinetic studies have not been conducted in the paediatric population.
Geriatric Patients
Ezetimibe
Plasma concentrations for total ezetimibe are about 2-fold higher in the elderly (≥65 years) than in the young (18 to 45 years). LDL-C reduction and safety profile is comparable between elderly and young subjects treated with ezetimibe. Therefore, no dosage adjustment is necessary in the elderly.
Atorvastatin
Plasma concentrations of atorvastatin are higher (approximately 40% for Cmax and 30% for AUC) in healthy elderly subjects (age ≥65 years) than in young adults. Lipid effects are comparable to that seen in younger patient populations given equal doses of atorvastatin.
Gender
Ezetimibe
Plasma concentrations for total ezetimibe are slightly higher (<20%) in women than in men. LDL-C reduction and safety profile are comparable between men and women treated with ezetimibe. Therefore, no dosage adjustment is necessary on the basis of gender.
Atorvastatin
Plasma concentrations of atorvastatin in women differ (approximately 20% higher for Cmax and 10% lower for AUC) from those in men; however, there is no clinically significant difference in lipid effects with atorvastatin between men and women.
Race
There are no pharmacokinetic data on the co-administration of ezetimibe and atorvastatin in nonCaucasians.
Ezetimibe
Based on a meta-analysis of pharmacokinetic studies with ezetimibe, there were no pharmacokinetic differences between Blacks and Caucasians.
Hepatic Insufficiency
Ezetimibe
After a single 10 mg dose of ezetimibe, the mean area under the curve (AUC) for total ezetimibe was increased approximately 1.7-fold in patients with mild hepatic insufficiency (Child Pugh score 5 or 6), compared to healthy subjects. In a 14 day, multiple-dose study (10 mg daily) in patients with moderate hepatic insufficiency (Child Pugh score 7 to 9), the mean AUC for total ezetimibe was increased approximately 4-fold on day 1 and day 14 compared to healthy subjects. No dosage adjustment is necessary for patients with mild hepatic insufficiency. Due to the unknown effects of the increased exposure to ezetimibe in patients with moderate or severe (Child Pugh score >9) hepatic insufficiency, ezetimibe is not recommended in these patients (see Section 4.4 Special Warnings and Precautions for Use).
Atorvastatin
Plasma concentrations of atorvastatin are markedly increased (approximately 16-fold in Cmax and 11-fold in AUC) in patients with chronic alcoholic liver disease (Child Pugh B) (see Section 4.2 Dose and Method of Administration, Section 4.3 Contraindications and Section 4.4 Special Warnings and Precautions for Use).
Renal Insufficiency
Ezetimibe
After a single 10 mg dose of ezetimibe in patients with severe renal disease (n=8; mean CrCl ≤30 mL/min/1.73 m²), the mean AUC for total ezetimibe was increased approximately 1.5-fold, compared to healthy subjects (n=9). This result is not considered to be clinically significant. No dosage adjustment is necessary for renally impaired patients. An additional patient in this study (post-renal transplant and receiving multiple medications, including ciclosporin) had a 12-fold greater exposure to total ezetimibe.
Atorvastatin
Renal disease has no influence on the plasma concentrations or lipid effects of atorvastatin; thus, dose adjustment in patients with renal dysfunction is not necessary (see Section 4.2 Dose and Method of Administration).
Haemodialysis
Haemodialysis is not expected to significantly enhance clearance of atorvastatin since the drug is extensively bound to plasma proteins.
Despite the expected cholesterol changes, no cardiovascular benefit with atorvastatin has been demonstrated in haemodialysis patients. Ezetimibe/atorvastatin has not been studied in this population.
5.3. Preclinical safety data
Genotoxicity
Ezetimibe
Ezetimibe alone or in combination with a statin (simvastatin, lovastatin, pravastatin or atorvastatin) or fenofibrate did not cause gene mutation in bacteria or chromosomal damage in human peripheral lymphocytes or bone marrow cells in mice.
Atorvastatin
Atorvastatin did not demonstrate mutagenic or clastogenic potential in an appropriate battery of assays. It was negative in the Ames test with Salmonella typhimurium and Escherichia coli, and in the in vitro HGPRT forward mutation assay in Chinese hamster lung cells. Atorvastatin did not produce significant increases in chromosomal aberrations in the in vitro Chinese hamster lung cell assay and was negative in the in vivo mouse micronucleus test.
Carcinogenicity
Ezetimibe
Two year dietary studies with ezetimibe alone in mice and rats showed no evidence of carcinogenic potential. The highest ezetimibe dose (500 mg/kg/day) in mice corresponds to exposure levels of approximately 4 and ≥ 150 times the adult human exposure for ezetimibe and total ezetimibe, respectively, based on AUC. Exposures in rats at the highest dose (1500 mg/kg/day in males and 500 mg/kg/day in females) correspond to approximately 2 and 14 times the adult human exposure for ezetimibe and total ezetimibe respectively. There are no carcinogenicity studies with ezetimibe/statin or ezetimibe/fenofibrate combinations.
Atorvastatin
In a 2 year study in rats given 10, 30 or 100 mg/kg/day, the incidence of hepatocellular adenoma was marginally, although not significantly, increased in females at 100 mg/kg/day. The maximum dose used was 11 times higher than the highest human dose (80 mg/kg) based on AUC(0–24) values. In a 2 year study in mice given 100, 200, or 400 mg/kg, incidences of hepatocellular adenoma in males and hepatocellular carcinoma in females were increased at 400 mg/kg. The maximum dose used was 14 times higher than the highest human dose (80 mg/kg) based on AUC(0-24) values. Other HMG-CoA reductase inhibitors have been reported to induce hepatocellular tumours in mice and rats.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.