LUTRATE DEPOT Powder and solvent for prolonged-release suspension for injection Ref.[6974] Active ingredients: Leuprorelin
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2017 Publisher: Mercury Pharmaceuticals Limited, Capital House, 85 King William Street, London EC4N 7BL, United Kingdom
Pharmacodynamic properties
Pharmacotherapeutic group: Endocrine therapy. Hormones and related agents. Gonadotropin-releasing hormones analogues
ATC code: L02AE02
Mechanism of action
The chemical name of Leuprorelin acetate is 5-oxo-L-prolyl-L-histidyl-L-tryptophyl-L-seryl-L-tyrosyl-D-leucyl-L-leucyl-L- arginyl-L-prolyl-ethylamide.
Leuprorelin acetate is inactive when given orally due to poor membrane permeability and an almost complete inactivation by intestinal proteolytic enzymes.
Leuprorelin acetate has potent LHRH agonist properties when given during short-term and intermittent therapy, however, when administered in a continuous, nonpulsatile manner, LHRH analogs induce inhibition of gonadotropin secretion and suppression of testicular steroidogenesis.
Pharmacodynamic effects
Upon binding to pituitary LHRH receptors, leuprorelin acetate produces an initial increase in circulating levels of luteinizing hormone (LH) and follicle stimulating hormone (FSH), leading to an acute rise in levels of testosterone and dihydrotestosterone. However, within five to eight days after drug administration, LHRH analogs produce desensitization of the LHRH receptor complex and/or downregulation of the anterior pituitary gland. Due to the fact that there are fewer receptors on the cell surface, cellular stimulation is decreased, and less gonadotropin is synthesized and secreted. Eventually, after several weeks of LHRH agonist therapy, LH and FSH secretion is suppressed. As a result, Leydig cells in the testes cease to produce testosterone, and the serum testosterone concentration declines to a castration level (less than 0.5 ng/mL) in about two to four weeks after initiation of treatment.
Clinical efficacy and safety
In an open-label, multicenter, multiple dose clinical study of Lutrate 1 month Depot, 160 patients with prostate cancer and no previous systemic cancer therapy, hormonal therapy for the treatment of prostate cancer, previous prostatic surgery neither previous orchiectomy, were enrolled. The objectives were to determine the efficacy and safety of Lutrate 1 month Depot when given to prostate cancer patients who could benefit from androgen deprivation therapy. Lutrate 1 month Depot was administered intramuscularly in 6 monthly doses.
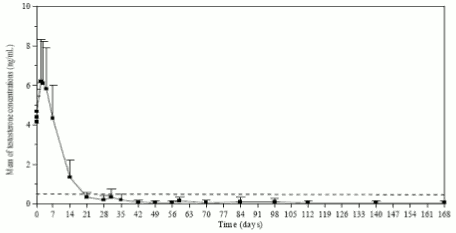
Testosterone levels were monitored at different days during 168 days. As expected, after the first injection the mean testosterone levels rapidly increased from baseline levels (4.119±1.341 ng/mL), reaching peak levels (Cmax) of 6.598±2.249 ng/mL at the third day. After peaking, testosterone levels fell, and by day 21, 78.7% of the evaluable patients had achieved medical castration (defined as testosterone less than 0.5 ng/mL). By Day 28, 96.8% of the patients had achieved castrate levels, and 73.1% had reached levels of ≤0.2 ng/mL (Figure 1).
Figure 1. Mean (±SD) testosterone plasma levels during treatment with six monthly IM injections of Lutrate 1 month Depot 3.75 mg:
Secondary efficacy endpoints included determination of serum LH, FSH and PSA concentrations. By day 14 and day 4 after the first Lutrate 1 month Depot injection, mean LH and FSH serum levels had decreased below the baseline concentrations. Concentrations remained well below baseline values from day 28 until the end of the study. During the treatment, mean PSA serum levels gradually decreased (first month) and then remained constantly below baseline level until the end of the study. However, a wide inter-individual variation in PSA concentrations was observed throughout the study.
The frequency of the acute on chronic response was 10.5% and the frequency of testosterone breakthrough response was 11.8%. No drug-related adverse events suggestive of a clinical testosterone flare (urinary retention, spinal cord compression, or exacerbation of bone pain) were reported in any of the patients showing a testosterone breakthrough effect.
Pharmacokinetic properties
Absorption
Following three once-monthly injections of Lutrate 1 month Depot in a sample of prostate cancer patients (N=12), maximal leuprorelin acetate plasma concentration was similar among the three cycles. After first administration (Days 0-28), Cmax was 13,145.6±3,070.6 pg/ml. Median time to achieve Cmax (Tmax) was 0.04 days, corresponding to 0.96 h (range 0.96–4.08 h).
Distribution
No drug distribution study was conducted with Lutrate 1 month Depot. However, in healthy male volunteers, the mean steady-state volume of distribution of leuprorelin acetate following bolus intravenous (IV) 1.0 mg dose was 27 L. In vitro binding to human plasma proteins ranged from 43% to 49%.
Elimination
No drug metabolism or excretion study was conducted with Lutrate 1 month Depot.
Leuprorelin is expected to be metabolised to smaller inactive peptides that may be excreted or further catabolised.
In healthy male volunteers, a 1.0 mg bolus of leuprorelin acetate administered IV revealed that the mean systemic clearance was 7.6 L/h, with a terminal elimination half-life of approximately 3 hours based on a two compartment model.
Following administration of leuprorelin acetate to 3 patients, less than 5% of the dose was recovered as parent and M-I metabolite in the urine. Special Populations
Renal/hepatic impairment
The pharmacokinetics of the drug in hepatically and renally impaired patients has not been determined.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity and genotoxicity conducted with leuprorelin acetate.
As expected from its known pharmacological properties, non-clinical studies showed effects on the reproductive systems, which were reversible. In the reproductive toxicity studies, leuprorelin acetate did not show teratogenicity. However, embryotoxicity/lethality was observed in rabbits.
Carcinogenicity studies performed in rats with leuprorelin acetate administered subcutaneously (0.6 to 4 mg/kg/day), showed a dose-related increase in pituitary adenomas Furthermore a significant but not dose-related increase of pancreatic islet-cell adenomas in females and of testicular interstitial cell adenomas in males was observed the highest incidence was in the low dose group. Administration of leuprorelin acetate resulted in inhibition of the growth of certain hormone dependent tumours (prostatic tumours in Noble and Dunning male rats and DMBA-induced mammary tumours in female rats). No such effects were observed in carcinogenicity studies performed in mice. No carcinogenicity studies have been conducted with Lutrate 1 month Depot.
Studies with leuprorelin acetate showed that the product was not mutagenic in a set of in vitro and in vivo assays. No mutagenicity studies have been conducted with Lutrate 1 month Depot.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.