PROLASTIN-C Powder for solution for injection Ref.[27444] Active ingredients: Alfa1 antitrypsin
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Alpha1-PI deficiency is an autosomal, co-dominant, hereditary disorder characterized by low serum and lung levels of Alpha1-PI. Smoking is an important risk factor for the development of emphysema in patients with Alpha1-PI deficiency.(1,2) Because emphysema affects many, but not all individuals with the more severe genetic variants of Alpha1-PI deficiency, augmentation therapy with Alpha1-PI is indicated only in patients with severe Alpha1-PI deficiency who have clinically evident emphysema.
Only some Alpha1-PI alleles are associated with clinically apparent Alpha1-PI deficiency.(3,4) Approximately 95% of all severely deficient patients are homozygous for the PiZ allele.(4) Individuals with the PiZZ variant typically have serum Alpha1-PI levels less than 35% of the average normal level. Individuals with the Pi(null)(null) variant have undetectable Alpha1-PI protein in their serum. Individuals with these low serum Alpha1-PI levels, i.e., less than 11 µM, have a markedly increased risk for developing emphysema over their lifetimes. In addition, PiSZ individuals, whose serum Alpha1-PI levels range from approximately 9 to 23 µM,(5) are considered to have moderately increased risk for developing emphysema, regardless of whether their serum Alpha1-PI levels are above or below 11 µM.
Augmenting the levels of functional protease inhibitor by intravenous infusion is an approach to therapy for patients with Alpha1-PI deficiency. The intended theoretical goal is to provide protection to the lower respiratory tract by correcting the imbalance between neutrophil elastase and protease inhibitors. Whether augmentation therapy with any Alpha1-PI product actually protects the lower respiratory tract from progressive emphysematous changes has not been demonstrated in adequately powered, randomized controlled, clinical trials. Although the maintenance of blood serum levels of Alpha1-PI (antigenically measured) above 11 µM has been historically postulated to provide therapeutically relevant anti-neutrophil elastase protection,(6) this has not been proven. Individuals with severe Alpha1-PI deficiency have been shown to have increased neutrophil and neutrophil elastase concentrations in lung epithelial lining fluid compared to normal PiMM individuals, and some PiSZ individuals with Alpha1-PI above 11 µM have emphysema attributed to Alpha1-PI deficiency. These observations underscore the uncertainty regarding the appropriate therapeutic target serum level of Alpha1-PI during augmentation therapy.
The pathogenesis of emphysema is understood as described in the "protease-antiprotease imbalance" model. Alpha1-PI is understood to be the primary antiprotease in the lower respiratory tract, where it inhibits neutrophil elastase (NE). Normal healthy individuals produce sufficient Alpha1-PI to control the NE produced by activated neutrophils and are thus able to prevent inappropriate proteolysis of the lung tissue by NE. Conditions that increase neutrophil accumulation and activation in the lung, such as respiratory infection and smoking, will in turn increase levels of NE. However, individuals who are severely deficient in endogenous Alpha1-PI are unable to maintain an appropriate antiprotease defense, and, in addition, they have been shown to have increased lung epithelial lining fluid neutrophil and NE concentrations. Because of these factors, many (but not all) individuals who are severely deficient in endogenous Alpha1-PI are subject to more rapid proteolysis of the alveolar walls leading to chronic lung disease. PROLASTIN -C (Alpha1-Proteinase Inhibitor [Human]) serves as Alpha1-PI augmentation therapy in the patient population with severe Alpha1-PI deficiency and emphysema, acting to increase and maintain serum and lung epithelial lining fluid levels of Alpha1-PI.
12.2. Pharmacodynamics
Chronic augmentation therapy with the predecessor product, PROLASTIN (Alpha1-Proteinase Inhibitor [Human]), administered weekly at a dose of 60 mg/kg body weight, results in statistically significant increased levels of Alpha1-PI and functional anti-neutrophil elastase capacity in the epithelial lining fluid of the lower respiratory tract of the lung, as compared to levels prior to commencing therapy with PROLASTIN.(7) However, the clinical benefit of the increased levels at the recommended dose has not been demonstrated in adequately powered, randomized, controlled clinical trials for any Alpha1-PI product.
12.3. Pharmacokinetics
The crossover pharmacokinetic (PK) study was a randomized, double-blind trial comparing PROLASTIN-C to PROLASTIN conducted in 24 adult subjects age 40 to 72 with severe Alpha1-PI deficiency. Ten subjects were male and 14 subjects were female. Twelve subjects were randomized to each treatment sequence. All but one subject had the PiZZ genotype and the remaining subject had PiSZ. All subjects had received prior Alpha1-PI therapy with PROLASTIN for at least 1 month.
Study subjects were randomly assigned to receive either 60 mg/kg body weight of functional PROLASTIN-C or PROLASTIN weekly by intravenous infusion during the first 8-week treatment period. Following the last dose in the first 8-week treatment period, subjects underwent serial blood sampling for PK analysis and then crossed over to the alternate treatment for the second 8-week treatment period. Following the last treatment in the second 8-week treatment period, subjects underwent serial blood sampling for PK analysis. In addition, blood samples were drawn for trough levels before infusion at Weeks 6, 7, and 8, as well as before infusion at Weeks 14, 15, and 16.
In the 8-week open-label treatment phase that followed the crossover period, all subjects received 60 mg/kg body weight of functional PROLASTIN-C.
The pharmacokinetic parameters of Alpha1-PI in plasma, based on functional activity assays, showed comparability between PROLASTIN-C treatment and PROLASTIN treatment, as shown in Table 5.
Table 5. Pharmacokinetic Parameters of Alpha1-PI in Plasma:
| Treatment | AUC0-7 days (hr*mg/mL) Mean (%CV) | Cmax (mg/mL) Mean (%CV) | t1/2 (hr) Mean (%CV) |
|---|---|---|---|
| PROLASTIN-C (n=22 or 23) | 155.9 (17%) | 1.797 (10%) | 146.3 (16%) |
| PROLASTIN (n=22 or 23) | 152.4 (16%) | 1.848 (15%) | 139.3 (18%) |
The key pharmacokinetic parameter was the area under the plasma concentration-time curve (AUC0-7days) following 8 weeks of treatment with PROLASTIN-C or PROLASTIN. The 90% confidence interval (0.97-1.09) for the ratio of AUC0-7days for PROLASTIN-C and PROLASTIN indicated that the 2 products are pharmacokinetically equivalent. Figure 1 shows the concentration (functional activity) vs. time curves of Alpha1-PI after intravenous administration of PROLASTIN-C and PROLASTIN.
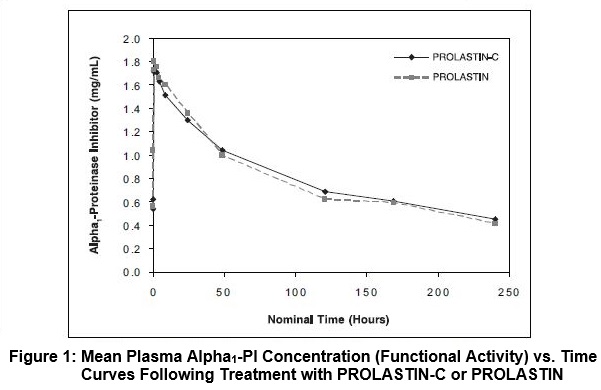
Trough levels measured during the crossover PK study via an antigenic content assay showed PROLASTIN-C treatment resulted in a mean trough of 16.9 ± 2.3 µM and PROLASTIN resulted in a mean trough of 16.7 ± 2.7 µM. Using the functional activity assay, PROLASTIN-C resulted in a mean trough of 11.8 ± 2.2 µM and PROLASTIN resulted in a mean trough of 11.0 ± 2.2 µM.
14. Clinical Studies
The clinical efficacy of PROLASTIN-C in influencing the course of pulmonary emphysema or pulmonary exacerbations has not been demonstrated in adequately powered, randomized, controlled clinical trials.
A total of 23 subjects with the PiZZ variant and documented emphysema were studied in a single-arm, open label clinical trial with PROLASTIN, the predecessor product. Nineteen of the subjects received PROLASTIN, 60 mg/kg, once weekly for up to 26 weeks (average 24 weeks). Blood levels of Alpha1-PI were maintained above 11 µM. Bronchoalveolar lavage studies demonstrated statistically significant increased levels of Alpha1-PI and functional ANEC in the epithelial lining fluid of the lower respiratory tract of the lung, as compared to levels prior to dosing.
A total of 62 individual subjects were studied in 2 clinical trials. In addition to the crossover pharmacokinetic study [see Clinical Pharmacology (12.3)], a multi-center, open-label single arm safety study was conducted to evaluate the safety and tolerability of PROLASTIN-C. In this study, 38 subjects were treated with weekly intravenous infusions of 60 mg/kg body weight of PROLASTIN-C for 20 weeks. Half the subjects were naïve to previous Alpha1-PI augmentation prior to study entry and the other half were receiving augmentation with PROLASTIN prior to entering the study. A diagnosis of severe Alpha1-PI deficiency was confirmed by the demonstration of the PiZZ genotype in 32 of 38 (84.2%) subjects, and 6 of 38 (15.8%) subjects presented with other alleles known to result in severe Alpha1-PI deficiency. These groups were distributed evenly between the naïve and non-naïve cohorts.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.